The Environmental Behaviour of Radium: Revised Edition the Environmental Behaviour of Radium: Revised
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
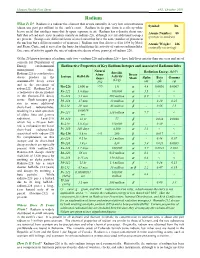
Radium What Is It? Radium Is a Radioactive Element That Occurs Naturally in Very Low Concentrations Symbol: Ra (About One Part Per Trillion) in the Earth’S Crust
Human Health Fact Sheet ANL, October 2001 Radium What Is It? Radium is a radioactive element that occurs naturally in very low concentrations Symbol: Ra (about one part per trillion) in the earth’s crust. Radium in its pure form is a silvery-white heavy metal that oxidizes immediately upon exposure to air. Radium has a density about one- Atomic Number: 88 half that of lead and exists in nature mainly as radium-226, although several additional isotopes (protons in nucleus) are present. (Isotopes are different forms of an element that have the same number of protons in the nucleus but a different number of neutrons.) Radium was first discovered in 1898 by Marie Atomic Weight: 226 and Pierre Curie, and it served as the basis for identifying the activity of various radionuclides. (naturally occurring) One curie of activity equals the rate of radioactive decay of one gram (g) of radium-226. Of the 25 known isotopes of radium, only two – radium-226 and radium-228 – have half-lives greater than one year and are of concern for Department of Energy environmental Radioactive Properties of Key Radium Isotopes and Associated Radionuclides management sites. Natural Specific Radiation Energy (MeV) Radium-226 is a radioactive Abun- Decay Isotope Half-Life Activity decay product in the dance Mode Alpha Beta Gamma (Ci/g) uranium-238 decay series (%) (α) (β) (γ) and is the precursor of Ra-226 1,600 yr >99 1.0 α 4.8 0.0036 0.0067 radon-222. Radium-228 is a radioactive decay product Rn-222 3.8 days 160,000 α 5.5 < < in the thorium-232 decay Po-218 3.1 min 290 million α 6.0 < < series. -

22Sra 226Ra and 222Rn in ' SOUTH CAROLINA GROUND WATER
Report No. 95 WRRI 22sRa 226Ra AND 222Rn IN ' SOUTH CAROLINA GROUND WATER: MEASUR~MEN-T TECHNIQUES AND ISOTOPE RELATIONSHIPS GlANNll\li FCUN~On o;: t,GR!CUL.TU~~tcoNOMlC2' ~~J.i\2Y J\li·iU ('lJ ~C 4· 1982 WATER RESOURCES RESEARCH INSTITUTE CLEMSON UNIVERSITY Clemson, .South Carolina JANUARY 1982 Water Resources Research Institute Clemson University Clemson, South Carolina 29631 ! 228Ra, 226Ra AND 222 Rn IN SOUTH CAROLINA GROUND WATER: MEASUREMENT TECHNIQUES AND ISOTOPE RELATIONSHIPS by Jacqueline Michel, Philip T. King and Willard S. Moore Department of Geology University of South Carolina Columbia, South Carolina 29208 The work upon which this puhlication is based was supported in part by funds provided by the Office of Water Research and Technology, project !!' No. OWRT-B-127-SC, U.S. Department of the Interior, fvashington, D. C., as authorized by the Water Research and Development Act of 1978 (PL95-467). r Project agreement No. 14-34-0001-9158 Period of Investigation: October 1979 - September 1980 Clemson University Water Resources Research Institute Technical Report No. 95 Contents of this publication do not necessarily reflect the views and policies of the Office of Water Research and Technology, U.S. Department of Interior, nor does mention of trade names or cormnercial products con stitute their endorsement or recommendation for use by the U.S. Goverrtment. ACKNOWLEDGEMENTS Much appreciation goes to Lewis Shaw, Rossie Stephens, Jim Ferguson and a large number of district personnel at the South Carolina Department of Health and Environmental Control who assisted us in field collection and provided much helpful information. Dr. -

Experimental Γ Ray Spectroscopy and Investigations of Environmental Radioactivity
Experimental γ Ray Spectroscopy and Investigations of Environmental Radioactivity BY RANDOLPH S. PETERSON 216 α Po 84 10.64h. 212 Pb 1- 415 82 0- 239 β- 01- 0 60.6m 212 1+ 1630 Bi 2+ 1513 83 α β- 2+ 787 304ns 0+ 0 212 α Po 84 Experimental γ Ray Spectroscopy and Investigations of Environmental Radioactivity Randolph S. Peterson Physics Department The University of the South Sewanee, Tennessee Published by Spectrum Techniques All Rights Reserved Copyright 1996 TABLE OF CONTENTS Page Introduction ....................................................................................................................4 Basic Gamma Spectroscopy 1. Energy Calibration ................................................................................................... 7 2. Gamma Spectra from Common Commercial Sources ........................................ 10 3. Detector Energy Resolution .................................................................................. 12 Interaction of Radiation with Matter 4. Compton Scattering............................................................................................... 14 5. Pair Production and Annihilation ........................................................................ 17 6. Absorption of Gammas by Materials ..................................................................... 19 7. X Rays ..................................................................................................................... 21 Radioactive Decay 8. Multichannel Scaling and Half-life ..................................................................... -

December 2020 Download
December 2020 Program Updates from VP and Program Director Doron Weber FILM Son of Monarchs Awarded 2021 Sloan Feature Film Prize at Sundance Son of Monarchs has been chosen as the Sloan Feature Film Prize winner at the 2021 Sundance Film Festival, to be awarded in January at this year’s virtual festival. The feature film, directed and written by filmmaker and scientist Alexis Gambis, follows a Mexican biologist who studies butterflies at a NYC lab as he returns home to the Monarch forests of Michoacán. The film was cited by the jury “for its poetic, multilayered portrait of a scientist’s growth and self-discovery as he migrates between Mexico and NYC working on transforming nature and uncovering the fluid boundaries that unite past and present and all living things.” This year’s Sloan jury included MIT researcher and protagonist of the new Sloan documentary Coded Bias Joy Buolamwini, Associate Professor in Chemistry at CUNY Hunter College and the CUNY Graduate Center Mandë Holford, and Sloan-supported filmmakers Aneesh Chaganty (Searching), Lydia Dean Pilcher (Radium Girls), and Lena Vurma (Adventures of a Mathematician). Son of Monarchs will be included in the 2021 Sundance Film Festival program and will be recognized at the closing night award ceremony as the Sloan winner. The Sloan Feature Film Prize, one of only six juried prizes at Sundance, is supported by a 2019 grant to the Sundance Institute. Ammonite Wins Sloan Science in Cinema Award at SFFILM This year’s SFFILM Sloan Science in Cinema Prize, which recognizes the compelling depiction of science in a narrative feature film, was awarded to Ammonite. -

Analysis of Isotopes of Radium, Uranium and Thorium. 210Pb-210Po
Analysis of isotopes of radium, uranium and thorium. 210Pb-210Po Per Roos Risø National Laboratory for Sustainable Energy, Technical university of Denmark Radium isotopes Adsorption of Ra on MnO2 & direct alpha spectrometry Coating of polyamid (Nylon) with MnO2 (KMnO4 solution) ……half-life of Ra-224 is 88h Very thin MnO2 layer, poor yield but excellent resolution Electrodeposited sources Direct 20 days 6 months Analysis of 226Ra in water • Weigh up a suitable amount of water (10 litre is mostly sufficient). • Add HNO3 to pH 1-2. • Add 133Ba tracer (ca 10 Bq). • Add about 13 ml of a 0.2M KMnO4. • Adjust pH to about 8-9 with ammonia. • Add 15 ml 0.3M MnCl2 and stir for some 5 minutes. • Adjust with KMnO4 or MnCl2 to make the sample water phase slightly pink. • Collect the precipitate and centrifuge • Dissolve precipitate in 1M HCl-1%H2O2. • Add about 2g K2SO4+1ml H2SO4 and dilute to 100 ml with water. • Heat the sample to dissolve the K2SO4 completely. • Add 30 mg Pb2+. Heat for 20 minutes and stir. • Centrifuge the precipitate and discard supernate. • Wash precipitate with 100 ml of a solution of 0.2M H2SO4 and 0.1 M K2SO4 • Centrifuge the precipitate and discard supernate • Dissolve the PbSO4-precipitate with 3ml 0.1M EDTA at pH 10. • Transfer the solution to the scintillation vial (low diffusion PE-vial). • Wash centrifuge tube with 2 ml 0.1M EDTA and transfer to Scintillation vial. • Add 10ml OptiFluor 0 and fill with deionised water to total 20 ml. • Allow for ingrowth of 222Rn during 3 weeks and measure by LSQ. -

Naturally Occurring Radioactive Material (NORM VI) (NORM Material Radioactive Occurring Naturally Naturally Occurring Radioactive Material
Naturally Occurring Radioactive Material (NORM VI) Naturally Occurring Radioactive Material All minerals and raw materials contain radionuclides of natural origin. For (NORM VI) most human activities involving minerals and raw materials, the levels of exposure to these radionuclides are not significantly greater than normal background levels. However, certain work activities can give rise to signif- Proceedings icantly enhanced exposures that may need to be controlled by regulation. Material giving rise to these enhanced exposures has become known as of an International Symposium naturally occurring radioactive material (NORM). Historically, most regula- tory attention has been focused on the mining and processing of uranium Marrakesh, Morocco, 22–26 March 2010 ore, because such activities are a direct consequence of the radioactivity in the ore and form part of the nuclear fuel cycle. Over the past decade or two, however, more and more countries have introduced measures to reg- 1 ulate exposures arising from a wider range of natural sources. The NORM VI symposium, which was held in Marrakesh, Morocco, from 22–26 March 2010, provided an important opportunity to review the developments that had taken place during the three year period since the NORM V sym- posium in 2007. This period, which began with the publication of new radiation protection recommendations by the International Commission on Radiological Protection, was characterized by ongoing activities to revise international standards on radiation protection and safety. These Proceedings contain all 38 papers accepted for oral presentation and four rapporteur reports, as well as a summary that concludes with the main findings of the symposium. Text versions of 43 poster presentations are provided on a CD-ROM which accompanies these Proceedings. -

Radionuclides Notice of Data Availability Technical Support Document
Radionuclides Notice of Data Availability Technical Support Document March 2000 Targeting and Analysis Branch Standards and Risk Management Division Office of Ground Water and Drinking Water United States Environmental Protection Agency Prepared by: USEPA Office of Ground Water and Drinking Water in collaboration with USEPA Office of Indoor Air and Radiation United States Geological Survey Preface Under the authority of the Safe Drinking Water Act (SDWA) and its amendments, EPA sets drinking water standards for contaminants, which potentially pose a threat to public health via a public drinking water source. In 1976, EPA promulgated National Interim Primary Drinking Water Regulations (NIPDWR) for three categories of radionuclides: gross alpha emitters, radium 226 and 228 combined, and gross beta and photon emitters. The 1976 NIPDWRs for these radionuclides regulated: 1. gross alpha at 15 picoCuries per liter (pCi/L)(excluding radon and uranium); 2. radium-226 and radium-228 combined at 5 pCi/L; 3. and beta/photon emitters at a 4 millirem dose of radioactivity. The 1986 reauthorization of the Safe Drinking Water Act (SDWA) declared the interim standards for these radionuclides to be final National Primary Drinking Water Regulations (NPDWR). The 1986 amendment also required standards to be set as close to the maximum contaminant level goal (MCLG; the health goal) as possible. At that time, radionuclides did not have an MCLG. In 1991 (56 FR 33050), EPA proposed: < A Maximum Contaminant Level Goal (MCLG) of zero for all radionuclides; < A -

Human Radiosensitivity and Radiosusceptibility: What Are the Differences?
International Journal of Molecular Sciences Review Human Radiosensitivity and Radiosusceptibility: What Are the Differences? Laura El-Nachef 1, Joelle Al-Choboq 1, Juliette Restier-Verlet 1, Adeline Granzotto 1, Elise Berthel 1,2, Laurène Sonzogni 1,Mélanie L. Ferlazzo 1, Audrey Bouchet 1 , Pierre Leblond 3, Patrick Combemale 3, Stéphane Pinson 4, Michel Bourguignon 1,5 and Nicolas Foray 1,* 1 Inserm, U1296 unit, Radiation: Defense, Health and Environment, Centre Léon-Bérard, 28, rue Laennec, 69008 Lyon, France; [email protected] (L.E.-N.); [email protected] (J.A.-C.); Juliette.Restier–[email protected] (J.R.-V.); [email protected] (A.G.); [email protected] (E.B.); [email protected] (L.S.); [email protected] (M.L.F.); [email protected] (A.B.); [email protected] (M.B.) 2 Neolys Diagnostics, 67960 Entzheim, France 3 Centre Léon-Bérard, 28, rue Laennec, 69008 Lyon, France; [email protected] (P.L.); [email protected] (P.C.) 4 Hospices Civils de Lyon, Quai des Célestins, 69002 Lyon, France; [email protected] 5 Université Paris Saclay Versailles St Quentin en Yvelines, 78035 Versailles, France * Correspondence: [email protected]; Tel.: +33-4-78-78-28-28 Abstract: The individual response to ionizing radiation (IR) raises a number of medical, scientific, and societal issues. While the term “radiosensitivity” was used by the pioneers at the beginning Citation: El-Nachef, L.; Al-Choboq, of the 20st century to describe only the radiation-induced adverse tissue reactions related to cell J.; Restier-Verlet, J.; Granzotto, A.; death, a confusion emerged in the literature from the 1930s, as “radiosensitivity” was indifferently Berthel, E.; Sonzogni, L.; Ferlazzo, used to describe the toxic, cancerous, or aging effect of IR. -

Occurrence of Radium-224, Radium
Prepared in cooperation with the New Jersey Department of Environmental Protection Occurrence of Radium-224, Radium-226 and Radium-228 in Water from the Vincentown and Wenonah-Mount Laurel Aquifers, the Englishtown Aquifer System, and the Hornerstown and Red Bank Sands, Southwestern and South-Central New Jersey Scientific Investigations Report 2007-5064 U.S. Department of the Interior U.S. Geological Survey Version 1.1 Occurrence of Radium-224, Radium-226 and Radium-228 in Water from the Vincentown and Wenonah-Mount Laurel Aquifers, the Englishtown Aquifer System, and the Hornerstown and Red Bank Sands, Southwestern and South-Central New Jersey By Vincent T. dePaul and Zoltan Szabo Prepared in cooperation with the New Jersey Department of Environmental Protection Scientific Investigations Report 2007–5064 U.S. Department of the Interior U.S. Geological Survey U.S. Department of the Interior DIRK KEMPTHORNE, Secretary U.S. Geological Survey Mark D. Myers, Director U.S. Geological Survey, Reston, Virginia: 2007 For product and ordering information: World Wide Web: http://www.usgs.gov/pubprod Telephone: 1-888-ASK-USGS For more information on the USGS--the Federal source for science about the Earth, its natural and living resources, natural hazards, and the environment: World Wide Web: http://www.usgs.gov Telephone: 1-888-ASK-USGS Any use of trade, product, or firm names is for descriptive purposes only and does not imply endorsement by the U.S. Government. Although this report is in the public domain, permission must be secured from the individual copyright owners to reproduce any copyrighted materials contained within this report. -

Determination of Radium-228 in Natural Waters
Determination of Radium-228 in Natural Waters GEOLOGICAL SURVEY WATER-SUPPLY PAPER 1696-G Determination of Radium-228 in Natural Waters By JESSE O. JOHNSON RADIOCHEMICAL ANALYSIS OF WATER GEOLOGICAL SURVEY WATER-SUPPLY PAPER 1696-G UNITED STATES GOVERNMENT PRINTING OFFICE, WASHINGTON : 1971 UNITED STATES DEPARTMENT OF THE INTERIOR ROGERS C. B. MORTON, Secretary GEOLOGICAL SURVEY William T. Pecora, Director Library of Congress catalog-card No. 72-610150 For sale by the Superintendent of Documents, U.S. Government Printing Office' Washington, D.C. 2O4O2 Price 25 cents (paper cover) CONTENTS Page Abstract ____________-______-_----------------------------------- Gl Introduction _______________---__-_------------------------------ 1 Review of radium-228 methods and studies -------------------- 5 Collection of water samples for radium-228 analysis ------------ 9 Development of the radium-228 method ---------------------------- 10 Natural radioactive nuclide interference ----------------------- 10 Radium-228 standard preparation ---------------------------- 13 Standards from "old" thorium nitrate ------------------------ 14 Standards from "old" thorium or thorium oxide _____-._-_-_---- 15 Standards from recently processed thorium reagents ----------- 15 Discussion of the method __.__-_---_-.__-_-__-------------------- 16 Apparatus ___._..__._______________.____-_-__------------------- 18 Reagents __________________---____--___---__-----_-------------- 18 Procedure for analysis of radium-228 ----------------------------- 20 Calculation of results ___-_--__---_--___-----_-----_-------------- 22 Errors and precision _______--_-_______-_-_-_-_--_---------------- 24 References ___.__.________________________________-__----------- 25 ILLUSTRATIONS Page FIGURE 1. Chart of the natural radioactive decay series ------------ G3 2. Graph of decay of airborne radioactivity trapped by membrane filters -______-_-----__--_-___--_------.-_- 12 TABLES Page TABLE 1. Radioactive element content of Colorado Front Range streams ____________________________________________ G8 2. -

Radium Girls
Mass Media & Environmental Conflict -- Radium Girls The Radium Girls By Bill Kovarik Originally published as Chapter Eight of Mass Media and Environmental Conflict (Revised 2002) Introduction The Consumers League Radium Dangers The Lawsuit The Media Coverage Walter Lippmann and the New York Press The Settlement and Its Aftermath Conclusion Footnotes It was a little strange, Fryer said, that when she blew her nose, her handkerchief glowed in the dark. But everyone knew the stuff was harmless. The women even painted their nails and their teeth to surprise their boyfriends when the lights went out. Illustration from the New York World, May 20, 1928. Grace Fryer and the other women at the radium factory in Orange, New Jersey, naturally supposed that they were not being poisoned. It was a little strange, Fryer said, that when she blew her nose, her handkerchief glowed in the dark. But everyone knew the stuff was harmless. The women even painted their nails and their teeth to surprise their boyfriends when the lights went out. They all had a good laugh, then got back to work, painting a glow-in-the-dark radium compound on the dials of watches, clocks, altimeters and other instruments. Grace started working in the spring of 1917 with 70 other women in a large, dusty room filled with long tables. Racks of dials waiting to be painted sat next to each woman's chair. They mixed up glue, water and radium The Orange, N.J. radium dial factory in the powder into a glowing greenish-white paint, and carefully mid-1920s. -

The Radium Dial Painters: Workers’ Rights, Scientific Est Ting, and the Fight for Humane Treatment Elizabeth Richter
Hamline University DigitalCommons@Hamline Departmental Honors Projects College of Liberal Arts Spring 2018 The Radium Dial Painters: Workers’ Rights, Scientific esT ting, and the Fight for Humane Treatment Elizabeth Richter Follow this and additional works at: https://digitalcommons.hamline.edu/dhp Part of the History Commons Recommended Citation Richter, Elizabeth, "The Radium Dial Painters: Workers’ Rights, Scientific eT sting, and the Fight for Humane Treatment" (2018). Departmental Honors Projects. 74. https://digitalcommons.hamline.edu/dhp/74 This Honors Project is brought to you for free and open access by the College of Liberal Arts at DigitalCommons@Hamline. It has been accepted for inclusion in Departmental Honors Projects by an authorized administrator of DigitalCommons@Hamline. For more information, please contact [email protected], [email protected]. The Radium Dial Painters: Workers’ Rights, Scientific Testing, and the Fight for Humane Treatment Elizabeth Richter An Honors Thesis Submitted for partial fulfillment of the requirements for graduation with honors in History from Hamline University April 28, 2018 Abstract From the early 1910s through the Great Depression, the dial painting industry provided opportune jobs for young female workers. Dial painting jobs did not require many skills but were well-paying professions. These careers attracted many young women and girls to work there. However, unknown to the painters at the time, the radium that they were using to paint the dial faces was slowly poisoning them and would later cause major health defects. Many of these women that did not die directly from the radium developed various forms of cancer and radium poisoning, which led to many lawsuits.