Predicting PDZ Domain Mediated Protein Interactions from Structure Shirley Hui1,2, Xiang Xing1,2 and Gary D Bader1,2,3*
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Murine MPDZ-Linked Hydrocephalus Is Caused by Hyperpermeability of the Choroid Plexus
Thomas Jefferson University Jefferson Digital Commons Cardeza Foundation for Hematologic Research Sidney Kimmel Medical College 1-1-2019 Murine MPDZ-linked hydrocephalus is caused by hyperpermeability of the choroid plexus. Junning Yang Thomas Jefferson University Claire Simonneau Thomas Jefferson University Robert Kilker Thomas Jefferson University Laura Oakley ThomasFollow this Jeff anderson additional University works at: https://jdc.jefferson.edu/cardeza_foundation Part of the Medical Pathology Commons Matthew D. Byrne ThomasLet us Jeff knowerson Univ howersity access to this document benefits ouy Recommended Citation See next page for additional authors Yang, Junning; Simonneau, Claire; Kilker, Robert; Oakley, Laura; Byrne, Matthew D.; Nichtova, Zuzana; Stefanescu, Ioana; Pardeep-Kumar, Fnu; Tripathi, Sushil; Londin, Eric; Saugier-Veber, Pascale; Willard, Belinda; Thakur, Mathew; Pickup, Stephen; Ishikawa, Hiroshi; Schroten, Horst; Smeyne, Richard; and Horowitz, Arie, "Murine MPDZ-linked hydrocephalus is caused by hyperpermeability of the choroid plexus." (2019). Cardeza Foundation for Hematologic Research. Paper 49. https://jdc.jefferson.edu/cardeza_foundation/49 This Article is brought to you for free and open access by the Jefferson Digital Commons. The Jefferson Digital Commons is a service of Thomas Jefferson University's Center for Teaching and Learning (CTL). The Commons is a showcase for Jefferson books and journals, peer-reviewed scholarly publications, unique historical collections from the University archives, and teaching tools. The Jefferson Digital Commons allows researchers and interested readers anywhere in the world to learn about and keep up to date with Jefferson scholarship. This article has been accepted for inclusion in Cardeza Foundation for Hematologic Research by an authorized administrator of the Jefferson Digital Commons. For more information, please contact: [email protected]. -

Microarray Analysis of Gene Expression in the Ovarian Cancer Cell Line HO-8910 with Silencing of the ZNF217 Gene
MOLECULAR MEDICINE REPORTS 2: 851-855, 2009 851 Microarray analysis of gene expression in the ovarian cancer cell line HO-8910 with silencing of the ZNF217 gene GUIQIN SUN1, JINGXIA QIN1, YUWEN QIU1, YUNFEI GAO1, YANHONG YU1, QINKAI DENG2 and MEI ZHONG1 1Department of Obstetrics and Gynecology, Nanfang Hospital, Southern Medical University; 2Department of Bioinformatics, Southern Medical University, Guangzhou 510515, Guangdong, P.R. China Received April 30, 2009; Accepted July 7, 2009 DOI: 10.3892/mmr_00000183 Abstract. Zinc-finger protein 217 (ZNF217), which is over- late stages (stage III or IV) and have a 5-year survival rate of expressed during cancer progression, can promote tumor cell less than 30% (1). Ovarian cancer is a heterogeneous disease immortalization. To examine the function of ZNF217, a global with respect to its histopathology, molecular biology and expression profile was carried out using Affymetrix Gene clinical outcome. Though studies on the etiology of ovarian Chip analysis with HG-U133 plus 2.0 arrays in the ovarian cancer and susceptibility to the disease have shown that the cancer cell line HO-8910 after silencing of the ZNF217 gene. mechanisms of carcinogenesis and cancer development are The results were analyzed using the Gene Ontology program associated with genetic, epigenetic and environmental factors, to investigate the functional network affected by ZNF217 the pathogenesis of ovarian cancer remains unclear (2). in ovarian cancer cells. Changes in the mRNA expression Among the current methods for the identification of gene of the affected genes were confirmed by real-time reverse expression profiles in cancer, microarray analysis is the most transcriptase-polymerase chain reaction (RT-PCR). -

The Utility of Genetic Risk Scores in Predicting the Onset of Stroke March 2021 6
DOT/FAA/AM-21/24 Office of Aerospace Medicine Washington, DC 20591 The Utility of Genetic Risk Scores in Predicting the Onset of Stroke Diana Judith Monroy Rios, M.D1 and Scott J. Nicholson, Ph.D.2 1. KR 30 # 45-03 University Campus, Building 471, 5th Floor, Office 510 Bogotá D.C. Colombia 2. FAA Civil Aerospace Medical Institute, 6500 S. MacArthur Blvd Rm. 354, Oklahoma City, OK 73125 March 2021 NOTICE This document is disseminated under the sponsorship of the U.S. Department of Transportation in the interest of information exchange. The United States Government assumes no liability for the contents thereof. _________________ This publication and all Office of Aerospace Medicine technical reports are available in full-text from the Civil Aerospace Medical Institute’s publications Web site: (www.faa.gov/go/oamtechreports) Technical Report Documentation Page 1. Report No. 2. Government Accession No. 3. Recipient's Catalog No. DOT/FAA/AM-21/24 4. Title and Subtitle 5. Report Date March 2021 The Utility of Genetic Risk Scores in Predicting the Onset of Stroke 6. Performing Organization Code 7. Author(s) 8. Performing Organization Report No. Diana Judith Monroy Rios M.D1, and Scott J. Nicholson, Ph.D.2 9. Performing Organization Name and Address 10. Work Unit No. (TRAIS) 1 KR 30 # 45-03 University Campus, Building 471, 5th Floor, Office 510, Bogotá D.C. Colombia 11. Contract or Grant No. 2 FAA Civil Aerospace Medical Institute, 6500 S. MacArthur Blvd Rm. 354, Oklahoma City, OK 73125 12. Sponsoring Agency name and Address 13. Type of Report and Period Covered Office of Aerospace Medicine Federal Aviation Administration 800 Independence Ave., S.W. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Circular RNA Hsa Circ 0005114‑Mir‑142‑3P/Mir‑590‑5P‑ Adenomatous
ONCOLOGY LETTERS 21: 58, 2021 Circular RNA hsa_circ_0005114‑miR‑142‑3p/miR‑590‑5p‑ adenomatous polyposis coli protein axis as a potential target for treatment of glioma BO WEI1*, LE WANG2* and JINGWEI ZHAO1 1Department of Neurosurgery, China‑Japan Union Hospital of Jilin University, Changchun, Jilin 130033; 2Department of Ophthalmology, The First Hospital of Jilin University, Jilin University, Changchun, Jilin 130021, P.R. China Received September 12, 2019; Accepted October 22, 2020 DOI: 10.3892/ol.2020.12320 Abstract. Glioma is the most common type of brain tumor APC expression with a good overall survival rate. UALCAN and is associated with a high mortality rate. Despite recent analysis using TCGA data of glioblastoma multiforme and the advances in treatment options, the overall prognosis in patients GSE25632 and GSE103229 microarray datasets showed that with glioma remains poor. Studies have suggested that circular hsa‑miR‑142‑3p/hsa‑miR‑590‑5p was upregulated and APC (circ)RNAs serve important roles in the development and was downregulated. Thus, hsa‑miR‑142‑3p/hsa‑miR‑590‑5p‑ progression of glioma and may have potential as therapeutic APC‑related circ/ceRNA axes may be important in glioma, targets. However, the expression profiles of circRNAs and their and hsa_circ_0005114 interacted with both of these miRNAs. functions in glioma have rarely been studied. The present study Functional analysis showed that hsa_circ_0005114 was aimed to screen differentially expressed circRNAs (DECs) involved in insulin secretion, while APC was associated with between glioma and normal brain tissues using sequencing the Wnt signaling pathway. In conclusion, hsa_circ_0005114‑ data collected from the Gene Expression Omnibus database miR‑142‑3p/miR‑590‑5p‑APC ceRNA axes may be potential (GSE86202 and GSE92322 datasets) and explain their mecha‑ targets for the treatment of glioma. -

Blood-Based Biomarkers for Predicting the Risk for Five-Year Incident Coronary Heart Disease in the Framingham Heart Study Via Machine Learning
G C A T T A C G G C A T genes Article Blood-Based Biomarkers for Predicting the Risk for Five-Year Incident Coronary Heart Disease in the Framingham Heart Study via Machine Learning Meeshanthini V. Dogan 1,2,3,*, Steven R. H. Beach 4, Ronald L. Simons 5, Amaury Lendasse 6,7, Brandan Penaluna 8 and Robert A. Philibert 1,2,3,8 1 Department of Biomedical Engineering, University of Iowa, Iowa City, IA 52242, USA; [email protected] 2 Cardio Diagnostics LLC, 2500 Crosspark Road, Coralville, IA 52241, USA 3 Department of Psychiatry, University of Iowa, Iowa City, IA 52242, USA 4 Department of Psychology, University of Georgia, Athens, GA 30602, USA; [email protected] 5 Department of Sociology, University of Georgia, Athens, GA 30606, USA; [email protected] 6 Information and Logistics Technology Department, University of Houston, Houston, TX 77004, USA; [email protected] 7 Department of Business Management and Analytics, Arcada University of Applied Sciences, 00560 Helsinki, Finland 8 Behavioral Diagnostics LLC, 2500 Crosspark Road, Coralville, IA 52241, USA; [email protected] * Correspondence: [email protected]; Tel.: +1-319-353-4986 Received: 15 November 2018; Accepted: 12 December 2018; Published: 18 December 2018 Abstract: An improved approach for predicting the risk for incident coronary heart disease (CHD) could lead to substantial improvements in cardiovascular health. Previously, we have shown that genetic and epigenetic loci could predict CHD status more sensitively than conventional risk factors. Herein, we examine whether similar machine learning approaches could be used to develop a similar panel for predicting incident CHD. -

1 UST College of Science Department of Biological Sciences
UST College of Science Department of Biological Sciences 1 Pharmacogenomics of Myofascial Pain Syndrome An Undergraduate Thesis Submitted to the Department of Biological Sciences College of Science University of Santo Tomas In Partial Fulfillment of the Requirements for the Degree of Bachelor of Science in Biology Jose Marie V. Lazaga Marc Llandro C. Fernandez May 2021 UST College of Science Department of Biological Sciences 2 PANEL APPROVAL SHEET This undergraduate research manuscript entitled: Pharmacogenomics of Myofascial Pain Syndrome prepared and submitted by Jose Marie V. Lazaga and Marc Llandro C. Fernandez, was checked and has complied with the revisions and suggestions requested by panel members after thorough evaluation. This final version of the manuscript is hereby approved and accepted for submission in partial fulfillment of the requirements for the degree of Bachelor of Science in Biology. Noted by: Asst. Prof. Marilyn G. Rimando, PhD Research adviser, Bio/MicroSem 602-603 Approved by: Bio/MicroSem 603 panel member Bio/MicroSem 603 panel member Date: Date: UST College of Science Department of Biological Sciences 3 DECLARATION OF ORIGINALITY We hereby affirm that this submission is our own work and that, to the best of our knowledge and belief, it contains no material previously published or written by another person nor material to which a substantial extent has been accepted for award of any other degree or diploma of a university or other institute of higher learning, except where due acknowledgement is made in the text. We also declare that the intellectual content of this undergraduate research is the product of our work, even though we may have received assistance from others on style, presentation, and language expression. -

1 Supporting Information for a Microrna Network Regulates
Supporting Information for A microRNA Network Regulates Expression and Biosynthesis of CFTR and CFTR-ΔF508 Shyam Ramachandrana,b, Philip H. Karpc, Peng Jiangc, Lynda S. Ostedgaardc, Amy E. Walza, John T. Fishere, Shaf Keshavjeeh, Kim A. Lennoxi, Ashley M. Jacobii, Scott D. Rosei, Mark A. Behlkei, Michael J. Welshb,c,d,g, Yi Xingb,c,f, Paul B. McCray Jr.a,b,c Author Affiliations: Department of Pediatricsa, Interdisciplinary Program in Geneticsb, Departments of Internal Medicinec, Molecular Physiology and Biophysicsd, Anatomy and Cell Biologye, Biomedical Engineeringf, Howard Hughes Medical Instituteg, Carver College of Medicine, University of Iowa, Iowa City, IA-52242 Division of Thoracic Surgeryh, Toronto General Hospital, University Health Network, University of Toronto, Toronto, Canada-M5G 2C4 Integrated DNA Technologiesi, Coralville, IA-52241 To whom correspondence should be addressed: Email: [email protected] (M.J.W.); yi- [email protected] (Y.X.); Email: [email protected] (P.B.M.) This PDF file includes: Materials and Methods References Fig. S1. miR-138 regulates SIN3A in a dose-dependent and site-specific manner. Fig. S2. miR-138 regulates endogenous SIN3A protein expression. Fig. S3. miR-138 regulates endogenous CFTR protein expression in Calu-3 cells. Fig. S4. miR-138 regulates endogenous CFTR protein expression in primary human airway epithelia. Fig. S5. miR-138 regulates CFTR expression in HeLa cells. Fig. S6. miR-138 regulates CFTR expression in HEK293T cells. Fig. S7. HeLa cells exhibit CFTR channel activity. Fig. S8. miR-138 improves CFTR processing. Fig. S9. miR-138 improves CFTR-ΔF508 processing. Fig. S10. SIN3A inhibition yields partial rescue of Cl- transport in CF epithelia. -

Genome-Wide DNA Methylation Analysis of KRAS Mutant Cell Lines Ben Yi Tew1,5, Joel K
www.nature.com/scientificreports OPEN Genome-wide DNA methylation analysis of KRAS mutant cell lines Ben Yi Tew1,5, Joel K. Durand2,5, Kirsten L. Bryant2, Tikvah K. Hayes2, Sen Peng3, Nhan L. Tran4, Gerald C. Gooden1, David N. Buckley1, Channing J. Der2, Albert S. Baldwin2 ✉ & Bodour Salhia1 ✉ Oncogenic RAS mutations are associated with DNA methylation changes that alter gene expression to drive cancer. Recent studies suggest that DNA methylation changes may be stochastic in nature, while other groups propose distinct signaling pathways responsible for aberrant methylation. Better understanding of DNA methylation events associated with oncogenic KRAS expression could enhance therapeutic approaches. Here we analyzed the basal CpG methylation of 11 KRAS-mutant and dependent pancreatic cancer cell lines and observed strikingly similar methylation patterns. KRAS knockdown resulted in unique methylation changes with limited overlap between each cell line. In KRAS-mutant Pa16C pancreatic cancer cells, while KRAS knockdown resulted in over 8,000 diferentially methylated (DM) CpGs, treatment with the ERK1/2-selective inhibitor SCH772984 showed less than 40 DM CpGs, suggesting that ERK is not a broadly active driver of KRAS-associated DNA methylation. KRAS G12V overexpression in an isogenic lung model reveals >50,600 DM CpGs compared to non-transformed controls. In lung and pancreatic cells, gene ontology analyses of DM promoters show an enrichment for genes involved in diferentiation and development. Taken all together, KRAS-mediated DNA methylation are stochastic and independent of canonical downstream efector signaling. These epigenetically altered genes associated with KRAS expression could represent potential therapeutic targets in KRAS-driven cancer. Activating KRAS mutations can be found in nearly 25 percent of all cancers1. -
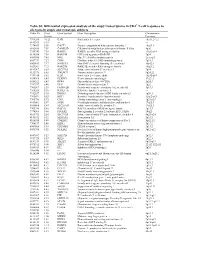
Table S1| Differential Expression Analysis of the Atopy Transcriptome
Table S1| Differential expression analysis of the atopy transcriptome in CD4+ T-cell responses to allergens in atopic and nonatopic subjects Probe ID S.test Gene Symbol Gene Description Chromosome Statistic Location 7994280 10.32 IL4R Interleukin 4 receptor 16p11.2-12.1 8143383 8.95 --- --- --- 7974689 8.50 DACT1 Dapper, antagonist of beta-catenin, homolog 1 14q23.1 8102415 7.59 CAMK2D Calcium/calmodulin-dependent protein kinase II delta 4q26 7950743 7.58 RAB30 RAB30, member RAS oncogene family 11q12-q14 8136580 7.54 RAB19B GTP-binding protein RAB19B 7q34 8043504 7.45 MAL Mal, T-cell differentiation protein 2cen-q13 8087739 7.27 CISH Cytokine inducible SH2-containing protein 3p21.3 8000413 7.17 NSMCE1 Non-SMC element 1 homolog (S. cerevisiae) 16p12.1 8021301 7.15 RAB27B RAB27B, member RAS oncogene family 18q21.2 8143367 6.83 SLC37A3 Solute carrier family 37 member 3 7q34 8152976 6.65 TMEM71 Transmembrane protein 71 8q24.22 7931914 6.56 IL2R Interleukin 2 receptor, alpha 10p15-p14 8014768 6.43 PLXDC1 Plexin domain containing 1 17q21.1 8056222 6.43 DPP4 Dipeptidyl-peptidase 4 (CD26) 2q24.3 7917697 6.40 GFI1 Growth factor independent 1 1p22 7903507 6.39 FAM102B Family with sequence similarity 102, member B 1p13.3 7968236 5.96 RASL11A RAS-like, family 11, member A --- 7912537 5.95 DHRS3 Dehydrogenase/reductase (SDR family) member 3 1p36.1 7963491 5.83 KRT1 Keratin 1 (epidermolytic hyperkeratosis) 12q12-q13 7903786 5.72 CSF1 Colony stimulating factor 1 (macrophage) 1p21-p13 8019061 5.67 SGSH N-sulfoglucosamine sulfohydrolase (sulfamidase) 17q25.3 -

Individual Protomers of a G Protein-Coupled Receptor Dimer Integrate Distinct Functional Modules
OPEN Citation: Cell Discovery (2015) 1, 15011; doi:10.1038/celldisc.2015.11 © 2015 SIBS, CAS All rights reserved 2056-5968/15 ARTICLE www.nature.com/celldisc Individual protomers of a G protein-coupled receptor dimer integrate distinct functional modules Nathan D Camp1, Kyung-Soon Lee2, Jennifer L Wacker-Mhyre2, Timothy S Kountz2, Ji-Min Park2, Dorathy-Ann Harris2, Marianne Estrada2, Aaron Stewart2, Alejandro Wolf-Yadlin1, Chris Hague2 1Department of Genome Sciences, University of Washington School of Medicine, Seattle, WA, USA; 2Department of Pharmacology, University of Washington School of Medicine, Seattle, WA, USA Recent advances in proteomic technology reveal G-protein-coupled receptors (GPCRs) are organized as large, macromolecular protein complexes in cell membranes, adding a new layer of intricacy to GPCR signaling. We previously reported the α1D-adrenergic receptor (ADRA1D)—a key regulator of cardiovascular, urinary and CNS function—binds the syntrophin family of PDZ domain proteins (SNTA, SNTB1, and SNTB2) through a C-terminal PDZ ligand inter- action, ensuring receptor plasma membrane localization and G-protein coupling. To assess the uniqueness of this novel GPCR complex, 23 human GPCRs containing Type I PDZ ligands were subjected to TAP/MS proteomic analysis. Syntrophins did not interact with any other GPCRs. Unexpectedly, a second PDZ domain protein, scribble (SCRIB), was detected in ADRA1D complexes. Biochemical, proteomic, and dynamic mass redistribution analyses indicate syntrophins and SCRIB compete for the PDZ ligand, simultaneously exist within an ADRA1D multimer, and impart divergent pharmacological properties to the complex. Our results reveal an unprecedented modular dimeric architecture for the ADRA1D in the cell membrane, providing unexpected opportunities for fine-tuning receptor function through novel protein interactions in vivo, and for intervening in signal transduction with small molecules that can stabilize or disrupt unique GPCR:PDZ protein interfaces. -

Aneuploidy: Using Genetic Instability to Preserve a Haploid Genome?
Health Science Campus FINAL APPROVAL OF DISSERTATION Doctor of Philosophy in Biomedical Science (Cancer Biology) Aneuploidy: Using genetic instability to preserve a haploid genome? Submitted by: Ramona Ramdath In partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biomedical Science Examination Committee Signature/Date Major Advisor: David Allison, M.D., Ph.D. Academic James Trempe, Ph.D. Advisory Committee: David Giovanucci, Ph.D. Randall Ruch, Ph.D. Ronald Mellgren, Ph.D. Senior Associate Dean College of Graduate Studies Michael S. Bisesi, Ph.D. Date of Defense: April 10, 2009 Aneuploidy: Using genetic instability to preserve a haploid genome? Ramona Ramdath University of Toledo, Health Science Campus 2009 Dedication I dedicate this dissertation to my grandfather who died of lung cancer two years ago, but who always instilled in us the value and importance of education. And to my mom and sister, both of whom have been pillars of support and stimulating conversations. To my sister, Rehanna, especially- I hope this inspires you to achieve all that you want to in life, academically and otherwise. ii Acknowledgements As we go through these academic journeys, there are so many along the way that make an impact not only on our work, but on our lives as well, and I would like to say a heartfelt thank you to all of those people: My Committee members- Dr. James Trempe, Dr. David Giovanucchi, Dr. Ronald Mellgren and Dr. Randall Ruch for their guidance, suggestions, support and confidence in me. My major advisor- Dr. David Allison, for his constructive criticism and positive reinforcement.