QUANTITATIVE TRAIT LOCUS ANALYSIS: in Several Variants (I.E., Alleles)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Murine MPDZ-Linked Hydrocephalus Is Caused by Hyperpermeability of the Choroid Plexus
Thomas Jefferson University Jefferson Digital Commons Cardeza Foundation for Hematologic Research Sidney Kimmel Medical College 1-1-2019 Murine MPDZ-linked hydrocephalus is caused by hyperpermeability of the choroid plexus. Junning Yang Thomas Jefferson University Claire Simonneau Thomas Jefferson University Robert Kilker Thomas Jefferson University Laura Oakley ThomasFollow this Jeff anderson additional University works at: https://jdc.jefferson.edu/cardeza_foundation Part of the Medical Pathology Commons Matthew D. Byrne ThomasLet us Jeff knowerson Univ howersity access to this document benefits ouy Recommended Citation See next page for additional authors Yang, Junning; Simonneau, Claire; Kilker, Robert; Oakley, Laura; Byrne, Matthew D.; Nichtova, Zuzana; Stefanescu, Ioana; Pardeep-Kumar, Fnu; Tripathi, Sushil; Londin, Eric; Saugier-Veber, Pascale; Willard, Belinda; Thakur, Mathew; Pickup, Stephen; Ishikawa, Hiroshi; Schroten, Horst; Smeyne, Richard; and Horowitz, Arie, "Murine MPDZ-linked hydrocephalus is caused by hyperpermeability of the choroid plexus." (2019). Cardeza Foundation for Hematologic Research. Paper 49. https://jdc.jefferson.edu/cardeza_foundation/49 This Article is brought to you for free and open access by the Jefferson Digital Commons. The Jefferson Digital Commons is a service of Thomas Jefferson University's Center for Teaching and Learning (CTL). The Commons is a showcase for Jefferson books and journals, peer-reviewed scholarly publications, unique historical collections from the University archives, and teaching tools. The Jefferson Digital Commons allows researchers and interested readers anywhere in the world to learn about and keep up to date with Jefferson scholarship. This article has been accepted for inclusion in Cardeza Foundation for Hematologic Research by an authorized administrator of the Jefferson Digital Commons. For more information, please contact: [email protected]. -

Lineage-Specific Mapping of Quantitative Trait Loci
Heredity (2013) 111, 106–113 & 2013 Macmillan Publishers Limited All rights reserved 0018-067X/13 www.nature.com/hdy ORIGINAL ARTICLE Lineage-specific mapping of quantitative trait loci C Chen1 and K Ritland We present an approach for quantitative trait locus (QTL) mapping, termed as ‘lineage-specific QTL mapping’, for inferring allelic changes of QTL evolution along with branches in a phylogeny. We describe and analyze the simplest case: by adding a third taxon into the normal procedure of QTL mapping between pairs of taxa, such inferences can be made along lineages to a presumed common ancestor. Although comparisons of QTL maps among species can identify homology of QTLs by apparent co- location, lineage-specific mapping of QTL can classify homology into (1) orthology (shared origin of QTL) versus (2) paralogy (independent origin of QTL within resolution of map distance). In this light, we present a graphical method that identifies six modes of QTL evolution in a three taxon comparison. We then apply our model to map lineage-specific QTLs for inbreeding among three taxa of yellow monkey-flower: Mimulus guttatus and two inbreeders M. platycalyx and M. micranthus, but critically assuming outcrossing was the ancestral state. The two most common modes of homology across traits were orthologous (shared ancestry of mutation for QTL alleles). The outbreeder M. guttatus had the fewest lineage-specific QTL, in accordance with the presumed ancestry of outbreeding. Extensions of lineage-specific QTL mapping to other types of data and crosses, and to inference of ancestral QTL state, are discussed. Heredity (2013) 111, 106–113; doi:10.1038/hdy.2013.24; published online 24 April 2013 Keywords: Quantitative trait locus mapping; evolution of mating systems; Mimulus INTRODUCTION common ancestor of these two taxa, back to the common ancestor of Knowledge of the genetic architecture of complex traits can offer all three taxa and forwards again to the third taxon. -
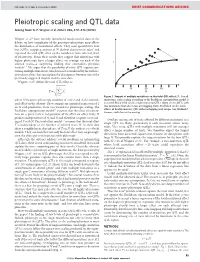
Pleiotropic Scaling and QTL Data Arising From: G
NATURE | Vol 456 | 4 December 2008 BRIEF COMMUNICATIONS ARISING Pleiotropic scaling and QTL data Arising from: G. P. Wagner et al. Nature 452, 470–473 (2008) Wagner et al.1 have recently introduced much-needed data to the debate on how complexity of the genotype–phenotype map affects the distribution of mutational effects. They used quantitative trait loci (QTLs) mapping analysis of 70 skeletal characters in mice2 and regressed the total QTL effect on the number of traits affected (level of pleiotropy). From their results they suggest that mutations with higher pleiotropy have a larger effect, on average, on each of the T affected traits—a surprising finding that contradicts previous models3–7. We argue that the possibility of some QTL regions con- taining multiple mutations, which was not considered by the authors, introduces a bias that can explain the discrepancy between one of the previously suggested models and the new data. Wagner et al.1 define the total QTL effect as vffiffiffiffiffiffiffiffiffiffiffiffiffi u uXN 10 20 30 ~t 2 T Ai N i~1 Figure 1 | Impact of multiple mutations on the total QTL effect, T. For all where N measures pleiotropy (number of traits) and Ai the normal- mutations, strict scaling according to the Euclidean superposition model is ized effect on the ith trait. They compare an empirical regression of T assumed. Black filled circles, single-mutation QTLs. Open circles, QTLs with on N with predictions from two models for pleiotropic scaling. The two mutations that affect non-overlapping traits. Red filled circles, total Euclidean superposition model3,4 assumes that the effect of a muta- effects of double-mutant QTL with overlapping trait ranges (see Methods). -

The Utility of Genetic Risk Scores in Predicting the Onset of Stroke March 2021 6
DOT/FAA/AM-21/24 Office of Aerospace Medicine Washington, DC 20591 The Utility of Genetic Risk Scores in Predicting the Onset of Stroke Diana Judith Monroy Rios, M.D1 and Scott J. Nicholson, Ph.D.2 1. KR 30 # 45-03 University Campus, Building 471, 5th Floor, Office 510 Bogotá D.C. Colombia 2. FAA Civil Aerospace Medical Institute, 6500 S. MacArthur Blvd Rm. 354, Oklahoma City, OK 73125 March 2021 NOTICE This document is disseminated under the sponsorship of the U.S. Department of Transportation in the interest of information exchange. The United States Government assumes no liability for the contents thereof. _________________ This publication and all Office of Aerospace Medicine technical reports are available in full-text from the Civil Aerospace Medical Institute’s publications Web site: (www.faa.gov/go/oamtechreports) Technical Report Documentation Page 1. Report No. 2. Government Accession No. 3. Recipient's Catalog No. DOT/FAA/AM-21/24 4. Title and Subtitle 5. Report Date March 2021 The Utility of Genetic Risk Scores in Predicting the Onset of Stroke 6. Performing Organization Code 7. Author(s) 8. Performing Organization Report No. Diana Judith Monroy Rios M.D1, and Scott J. Nicholson, Ph.D.2 9. Performing Organization Name and Address 10. Work Unit No. (TRAIS) 1 KR 30 # 45-03 University Campus, Building 471, 5th Floor, Office 510, Bogotá D.C. Colombia 11. Contract or Grant No. 2 FAA Civil Aerospace Medical Institute, 6500 S. MacArthur Blvd Rm. 354, Oklahoma City, OK 73125 12. Sponsoring Agency name and Address 13. Type of Report and Period Covered Office of Aerospace Medicine Federal Aviation Administration 800 Independence Ave., S.W. -

1 ROBERT PLOMIN on Behavioral Genetics Genetic Links Common
Genetic links • Background – Why has educational psychology ben so slow ROBERT PLOMIN to accept genetics? on Behavioral Genetics • Beyond nature vs. nurture – 3 findings from genetic research with far- reaching implications for education: October, 2004 The nature of nurture The abnormal is normal Generalist genes • Molecular Genetics Quantitative genetics: twin and adoption studies Common medical disorders Identical twins Fraternal twins (monozygotic, MZ) (dizygotic, DZ) 1 Twin concordances in TEDS at 7 years Twins Early Development Study (TEDS) for low reading and maths Perceptions of nature/nurture: % parents (N=1340) and teachers Beyond nature vs. nurture: (N=667) indicating that genes are at least as important as environment (Walker & Plomin, Ed. Psych., in press) The nature of nurture • Genetic influence on experience • Genotype-environment correlation – Children’s genetic propensities are correlated with their experiences passive evocative active 2 Beyond nature vs. nurture: Genotype-environment correlation The nature of nurture • Passive • Genotype-environment correlation – e.g., family background (SES) and school – Children’s genetic propensities are correlated with achievement their experiences passive • Evocative evocative – Children evoke experiences for genetic reasons Active • Active • Molecular genetics – Children actively create environments that foster – Find genes associated with learning experiences their genetic propensities Beyond nature vs. nurture: The one-gene one-disability (OGOD) model The abnormal is normal • Common learning disabilities are the quantitative extreme of the same genetic and environmental factors responsible for normal variation in learning abilities. • There are no common learning disabilities, just the extremes of normal variation. 3 OGOD (rare) and QTL (common) disorders The quantitative trait locus (QTL) model Beyond nature vs. -

Supplementary Table 1: Adhesion Genes Data Set
Supplementary Table 1: Adhesion genes data set PROBE Entrez Gene ID Celera Gene ID Gene_Symbol Gene_Name 160832 1 hCG201364.3 A1BG alpha-1-B glycoprotein 223658 1 hCG201364.3 A1BG alpha-1-B glycoprotein 212988 102 hCG40040.3 ADAM10 ADAM metallopeptidase domain 10 133411 4185 hCG28232.2 ADAM11 ADAM metallopeptidase domain 11 110695 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 195222 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 165344 8751 hCG20021.3 ADAM15 ADAM metallopeptidase domain 15 (metargidin) 189065 6868 null ADAM17 ADAM metallopeptidase domain 17 (tumor necrosis factor, alpha, converting enzyme) 108119 8728 hCG15398.4 ADAM19 ADAM metallopeptidase domain 19 (meltrin beta) 117763 8748 hCG20675.3 ADAM20 ADAM metallopeptidase domain 20 126448 8747 hCG1785634.2 ADAM21 ADAM metallopeptidase domain 21 208981 8747 hCG1785634.2|hCG2042897 ADAM21 ADAM metallopeptidase domain 21 180903 53616 hCG17212.4 ADAM22 ADAM metallopeptidase domain 22 177272 8745 hCG1811623.1 ADAM23 ADAM metallopeptidase domain 23 102384 10863 hCG1818505.1 ADAM28 ADAM metallopeptidase domain 28 119968 11086 hCG1786734.2 ADAM29 ADAM metallopeptidase domain 29 205542 11085 hCG1997196.1 ADAM30 ADAM metallopeptidase domain 30 148417 80332 hCG39255.4 ADAM33 ADAM metallopeptidase domain 33 140492 8756 hCG1789002.2 ADAM7 ADAM metallopeptidase domain 7 122603 101 hCG1816947.1 ADAM8 ADAM metallopeptidase domain 8 183965 8754 hCG1996391 ADAM9 ADAM metallopeptidase domain 9 (meltrin gamma) 129974 27299 hCG15447.3 ADAMDEC1 ADAM-like, -

Epigenome-Wide Study Identified Methylation Sites Associated With
nutrients Article Epigenome-Wide Study Identified Methylation Sites Associated with the Risk of Obesity Majid Nikpay 1,*, Sepehr Ravati 2, Robert Dent 3 and Ruth McPherson 1,4,* 1 Ruddy Canadian Cardiovascular Genetics Centre, University of Ottawa Heart Institute, 40 Ruskin St–H4208, Ottawa, ON K1Y 4W7, Canada 2 Plastenor Technologies Company, Montreal, QC H2P 2G4, Canada; [email protected] 3 Department of Medicine, Division of Endocrinology, University of Ottawa, the Ottawa Hospital, Ottawa, ON K1Y 4E9, Canada; [email protected] 4 Atherogenomics Laboratory, University of Ottawa Heart Institute, Ottawa, ON K1Y 4W7, Canada * Correspondence: [email protected] (M.N.); [email protected] (R.M.) Abstract: Here, we performed a genome-wide search for methylation sites that contribute to the risk of obesity. We integrated methylation quantitative trait locus (mQTL) data with BMI GWAS information through a SNP-based multiomics approach to identify genomic regions where mQTLs for a methylation site co-localize with obesity risk SNPs. We then tested whether the identified site contributed to BMI through Mendelian randomization. We identified multiple methylation sites causally contributing to the risk of obesity. We validated these findings through a replication stage. By integrating expression quantitative trait locus (eQTL) data, we noted that lower methylation at cg21178254 site upstream of CCNL1 contributes to obesity by increasing the expression of this gene. Higher methylation at cg02814054 increases the risk of obesity by lowering the expression of MAST3, whereas lower methylation at cg06028605 contributes to obesity by decreasing the expression of SLC5A11. Finally, we noted that rare variants within 2p23.3 impact obesity by making the cg01884057 Citation: Nikpay, M.; Ravati, S.; Dent, R.; McPherson, R. -

High Throughput Computational Mouse Genetic Analysis
bioRxiv preprint doi: https://doi.org/10.1101/2020.09.01.278465; this version posted January 22, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. High Throughput Computational Mouse Genetic Analysis Ahmed Arslan1+, Yuan Guan1+, Zhuoqing Fang1, Xinyu Chen1, Robin Donaldson2&, Wan Zhu, Madeline Ford1, Manhong Wu, Ming Zheng1, David L. Dill2* and Gary Peltz1* 1Department of Anesthesia, Stanford University School of Medicine, Stanford, CA; and 2Department of Computer Science, Stanford University, Stanford, CA +These authors contributed equally to this paper & Current Address: Ecree Durham, NC 27701 *Address correspondence to: [email protected] bioRxiv preprint doi: https://doi.org/10.1101/2020.09.01.278465; this version posted January 22, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Background: Genetic factors affecting multiple biomedical traits in mice have been identified when GWAS data that measured responses in panels of inbred mouse strains was analyzed using haplotype-based computational genetic mapping (HBCGM). Although this method was previously used to analyze one dataset at a time; but now, a vast amount of mouse phenotypic data is now publicly available, which could lead to many more genetic discoveries. Results: HBCGM and a whole genome SNP map covering 53 inbred strains was used to analyze 8462 publicly available datasets of biomedical responses (1.52M individual datapoints) measured in panels of inbred mouse strains. As proof of concept, causative genetic factors affecting susceptibility for eye, metabolic and infectious diseases were identified when structured automated methods were used to analyze the output. -

Transcriptome Analysis of Human Diabetic Kidney Disease
ORIGINAL ARTICLE Transcriptome Analysis of Human Diabetic Kidney Disease Karolina I. Woroniecka,1 Ae Seo Deok Park,1 Davoud Mohtat,2 David B. Thomas,3 James M. Pullman,4 and Katalin Susztak1,5 OBJECTIVE—Diabetic kidney disease (DKD) is the single cases, mild and then moderate mesangial expansion can be leading cause of kidney failure in the U.S., for which a cure has observed. In general, diabetic kidney disease (DKD) is not yet been found. The aim of our study was to provide an considered a nonimmune-mediated degenerative disease unbiased catalog of gene-expression changes in human diabetic of the glomerulus; however, it has long been noted that kidney biopsy samples. complement and immunoglobulins sometimes can be de- — tected in diseased glomeruli, although their role and sig- RESEARCH DESIGN AND METHODS Affymetrix expression fi arrays were used to identify differentially regulated transcripts in ni cance is not clear (4). 44 microdissected human kidney samples. The DKD samples were The understanding of DKD has been challenged by multi- significant for their racial diversity and decreased glomerular ple issues. First, the diagnosis of DKD usually is made using filtration rate (~20–30 mL/min). Stringent statistical analysis, using clinical criteria, and kidney biopsy often is not performed. the Benjamini-Hochberg corrected two-tailed t test, was used to According to current clinical practice, the development of identify differentially expressed transcripts in control and diseased albuminuria in patients with diabetes is sufficient to make the glomeruli and tubuli. Two different Web-based algorithms were fi diagnosis of DKD (5). We do not understand the correlation used to de ne differentially regulated pathways. -

Article the Power to Detect Quantitative Trait Loci Using
The Power to Detect Quantitative Trait Loci Using Resequenced, Experimentally Evolved Populations of Diploid, Sexual Organisms James G. Baldwin-Brown,*,1 Anthony D. Long,1 and Kevin R. Thornton1 1Department of Ecology and Evolutionary Biology, University of California, Irvine *Corresponding author: E-mail: [email protected]. Associate editor: John Parsch Abstract A novel approach for dissecting complex traits is to experimentally evolve laboratory populations under a controlled environment shift, resequence the resulting populations, and identify single nucleotide polymorphisms (SNPs) and/or genomic regions highly diverged in allele frequency. To better understand the power and localization ability of such an evolve and resequence (E&R) approach, we carried out forward-in-time population genetics simulations of 1 Mb genomic regions under a large combination of experimental conditions, then attempted to detect significantly diverged SNPs. Our analysis indicates that the ability to detect differentiation between populations is primarily affected by selection coef- ficient, population size, number of replicate populations, and number of founding haplotypes. We estimate that E&R studies can detect and localize causative sites with 80% success or greater when the number of founder haplotypes is over 500, experimental populations are replicated at least 25-fold, population size is at least 1,000 diploid individuals, and the selection coefficient on the locus of interest is at least 0.1. More achievable experimental designs (less replicated, fewer founder haplotypes, smaller effective population size, and smaller selection coefficients) can have power of greater than 50% to identify a handful of SNPs of which one is likely causative. Similarly, in cases where s 0.2, less demanding experimental designs can yield high power. -

Glossary/Index
Glossary 03/08/2004 9:58 AM Page 119 GLOSSARY/INDEX The numbers after each term represent the chapter in which it first appears. additive 2 allele 2 When an allele’s contribution to the variation in a One of two or more alternative forms of a gene; a single phenotype is separately measurable; the independent allele for each gene is inherited separately from each effects of alleles “add up.” Antonym of nonadditive. parent. ADHD/ADD 6 Alzheimer’s disease 5 Attention Deficit Hyperactivity Disorder/Attention A medical disorder causing the loss of memory, rea- Deficit Disorder. Neurobehavioral disorders character- soning, and language abilities. Protein residues called ized by an attention span or ability to concentrate that is plaques and tangles build up and interfere with brain less than expected for a person's age. With ADHD, there function. This disorder usually first appears in persons also is age-inappropriate hyperactivity, impulsive over age sixty-five. Compare to early-onset Alzheimer’s. behavior or lack of inhibition. There are several types of ADHD: a predominantly inattentive subtype, a predomi- amino acids 2 nantly hyperactive-impulsive subtype, and a combined Molecules that are combined to form proteins. subtype. The condition can be cognitive alone or both The sequence of amino acids in a protein, and hence pro- cognitive and behavioral. tein function, is determined by the genetic code. adoption study 4 amnesia 5 A type of research focused on families that include one Loss of memory, temporary or permanent, that can result or more children raised by persons other than their from brain injury, illness, or trauma. -

Genomic and Expression Profiling of Human Spermatocytic Seminomas: Primary Spermatocyte As Tumorigenic Precursor and DMRT1 As Candidate Chromosome 9 Gene
Research Article Genomic and Expression Profiling of Human Spermatocytic Seminomas: Primary Spermatocyte as Tumorigenic Precursor and DMRT1 as Candidate Chromosome 9 Gene Leendert H.J. Looijenga,1 Remko Hersmus,1 Ad J.M. Gillis,1 Rolph Pfundt,4 Hans J. Stoop,1 Ruud J.H.L.M. van Gurp,1 Joris Veltman,1 H. Berna Beverloo,2 Ellen van Drunen,2 Ad Geurts van Kessel,4 Renee Reijo Pera,5 Dominik T. Schneider,6 Brenda Summersgill,7 Janet Shipley,7 Alan McIntyre,7 Peter van der Spek,3 Eric Schoenmakers,4 and J. Wolter Oosterhuis1 1Department of Pathology, Josephine Nefkens Institute; Departments of 2Clinical Genetics and 3Bioinformatics, Erasmus Medical Center/ University Medical Center, Rotterdam, the Netherlands; 4Department of Human Genetics, Radboud University Medical Center, Nijmegen, the Netherlands; 5Howard Hughes Medical Institute, Whitehead Institute and Department of Biology, Massachusetts Institute of Technology, Cambridge, Massachusetts; 6Clinic of Paediatric Oncology, Haematology and Immunology, Heinrich-Heine University, Du¨sseldorf, Germany; 7Molecular Cytogenetics, Section of Molecular Carcinogenesis, The Institute of Cancer Research, Sutton, Surrey, United Kingdom Abstract histochemistry, DMRT1 (a male-specific transcriptional regulator) was identified as a likely candidate gene for Spermatocytic seminomas are solid tumors found solely in the involvement in the development of spermatocytic seminomas. testis of predominantly elderly individuals. We investigated these tumors using a genome-wide analysis for structural and (Cancer Res 2006; 66(1): 290-302) numerical chromosomal changes through conventional kar- yotyping, spectral karyotyping, and array comparative Introduction genomic hybridization using a 32 K genomic tiling-path Spermatocytic seminomas are benign testicular tumors that resolution BAC platform (confirmed by in situ hybridization).