100 Years of Quantitative Genetics Theory and Its Applications: Celebrating the Centenary of Fisher 1918”
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Lineage-Specific Mapping of Quantitative Trait Loci
Heredity (2013) 111, 106–113 & 2013 Macmillan Publishers Limited All rights reserved 0018-067X/13 www.nature.com/hdy ORIGINAL ARTICLE Lineage-specific mapping of quantitative trait loci C Chen1 and K Ritland We present an approach for quantitative trait locus (QTL) mapping, termed as ‘lineage-specific QTL mapping’, for inferring allelic changes of QTL evolution along with branches in a phylogeny. We describe and analyze the simplest case: by adding a third taxon into the normal procedure of QTL mapping between pairs of taxa, such inferences can be made along lineages to a presumed common ancestor. Although comparisons of QTL maps among species can identify homology of QTLs by apparent co- location, lineage-specific mapping of QTL can classify homology into (1) orthology (shared origin of QTL) versus (2) paralogy (independent origin of QTL within resolution of map distance). In this light, we present a graphical method that identifies six modes of QTL evolution in a three taxon comparison. We then apply our model to map lineage-specific QTLs for inbreeding among three taxa of yellow monkey-flower: Mimulus guttatus and two inbreeders M. platycalyx and M. micranthus, but critically assuming outcrossing was the ancestral state. The two most common modes of homology across traits were orthologous (shared ancestry of mutation for QTL alleles). The outbreeder M. guttatus had the fewest lineage-specific QTL, in accordance with the presumed ancestry of outbreeding. Extensions of lineage-specific QTL mapping to other types of data and crosses, and to inference of ancestral QTL state, are discussed. Heredity (2013) 111, 106–113; doi:10.1038/hdy.2013.24; published online 24 April 2013 Keywords: Quantitative trait locus mapping; evolution of mating systems; Mimulus INTRODUCTION common ancestor of these two taxa, back to the common ancestor of Knowledge of the genetic architecture of complex traits can offer all three taxa and forwards again to the third taxon. -
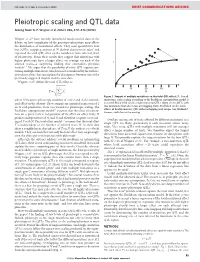
Pleiotropic Scaling and QTL Data Arising From: G
NATURE | Vol 456 | 4 December 2008 BRIEF COMMUNICATIONS ARISING Pleiotropic scaling and QTL data Arising from: G. P. Wagner et al. Nature 452, 470–473 (2008) Wagner et al.1 have recently introduced much-needed data to the debate on how complexity of the genotype–phenotype map affects the distribution of mutational effects. They used quantitative trait loci (QTLs) mapping analysis of 70 skeletal characters in mice2 and regressed the total QTL effect on the number of traits affected (level of pleiotropy). From their results they suggest that mutations with higher pleiotropy have a larger effect, on average, on each of the T affected traits—a surprising finding that contradicts previous models3–7. We argue that the possibility of some QTL regions con- taining multiple mutations, which was not considered by the authors, introduces a bias that can explain the discrepancy between one of the previously suggested models and the new data. Wagner et al.1 define the total QTL effect as vffiffiffiffiffiffiffiffiffiffiffiffiffi u uXN 10 20 30 ~t 2 T Ai N i~1 Figure 1 | Impact of multiple mutations on the total QTL effect, T. For all where N measures pleiotropy (number of traits) and Ai the normal- mutations, strict scaling according to the Euclidean superposition model is ized effect on the ith trait. They compare an empirical regression of T assumed. Black filled circles, single-mutation QTLs. Open circles, QTLs with on N with predictions from two models for pleiotropic scaling. The two mutations that affect non-overlapping traits. Red filled circles, total Euclidean superposition model3,4 assumes that the effect of a muta- effects of double-mutant QTL with overlapping trait ranges (see Methods). -

1 ROBERT PLOMIN on Behavioral Genetics Genetic Links Common
Genetic links • Background – Why has educational psychology ben so slow ROBERT PLOMIN to accept genetics? on Behavioral Genetics • Beyond nature vs. nurture – 3 findings from genetic research with far- reaching implications for education: October, 2004 The nature of nurture The abnormal is normal Generalist genes • Molecular Genetics Quantitative genetics: twin and adoption studies Common medical disorders Identical twins Fraternal twins (monozygotic, MZ) (dizygotic, DZ) 1 Twin concordances in TEDS at 7 years Twins Early Development Study (TEDS) for low reading and maths Perceptions of nature/nurture: % parents (N=1340) and teachers Beyond nature vs. nurture: (N=667) indicating that genes are at least as important as environment (Walker & Plomin, Ed. Psych., in press) The nature of nurture • Genetic influence on experience • Genotype-environment correlation – Children’s genetic propensities are correlated with their experiences passive evocative active 2 Beyond nature vs. nurture: Genotype-environment correlation The nature of nurture • Passive • Genotype-environment correlation – e.g., family background (SES) and school – Children’s genetic propensities are correlated with achievement their experiences passive • Evocative evocative – Children evoke experiences for genetic reasons Active • Active • Molecular genetics – Children actively create environments that foster – Find genes associated with learning experiences their genetic propensities Beyond nature vs. nurture: The one-gene one-disability (OGOD) model The abnormal is normal • Common learning disabilities are the quantitative extreme of the same genetic and environmental factors responsible for normal variation in learning abilities. • There are no common learning disabilities, just the extremes of normal variation. 3 OGOD (rare) and QTL (common) disorders The quantitative trait locus (QTL) model Beyond nature vs. -

Response to Selection in Finite Locus Models with Nonadditive Effects
Journal of Heredity, 2017, 318–327 doi:10.1093/jhered/esw123 Original Article Advance Access publication January 12, 2017 Original Article Response to Selection in Finite Locus Models with Nonadditive Effects Hadi Esfandyari, Mark Henryon, Peer Berg, Jørn Rind Thomasen, Downloaded from https://academic.oup.com/jhered/article/108/3/318/2895120 by guest on 23 September 2021 Piter Bijma, and Anders Christian Sørensen From the Center for Quantitative Genetics and Genomics, Department of Molecular Biology and Genetics, Aarhus University DK-8830 Tjele, Denmark (Esfandyari, Berg, Thomasen, and Sørensen); Seges, Danish Pig Research Centre, Copenhagen, Denmark (Henryon); School of Animal Biology, University of Western Australia, Crawley, Australia (Henryon); Nordic Genetic Resource Center, Ås, Norway (Berg); VikingGenetics, Assentoft, Denmark (Thomasen); and Animal Breeding and Genomics Centre, Wageningen University, Wageningen, The Netherlands (Bijma). Address correspondence to H. Esfandyari at the address above, or e-mail: hadi.esfandyari@ mbg.au.dk. Received May 18, 2016; First decision June 7, 2016; Accepted January 2, 2017. Corresponding Editor: C Scott Baker Abstract Under the finite-locus model in the absence of mutation, the additive genetic variation is expected to decrease when directional selection is acting on a population, according to quantitative-genetic theory. However, some theoretical studies of selection suggest that the level of additive variance can be sustained or even increased when nonadditive genetic effects are present. We tested the hypothesis that finite-locus models with both additive and nonadditive genetic effects maintain more additive genetic variance (VA) and realize larger medium- to long-term genetic gains than models with only additive effects when the trait under selection is subject to truncation selection. -

Directional Selection and Variation in Finite Populations
Copyright 0 1987 by the Genetics Society of America Directional Selection and Variation in Finite Populations Peter D. Keightley and ‘William G. Hill Institute of Animal Genetics, University of Edinburgh, West Mains Road, Edinburgh EH9 3JN, Scotland Manuscript received April 12, 1987 Revised copy accepted July 16, 1987 ABSTRACT Predictions are made of the equilibrium genetic variances and responses in a metric trait under the joint effects of directional selection, mutation and linkage in a finite population. The “infinitesimal model“ is analyzed as the limiting case of many mutants of very small effect, otherwise Monte Carlo simulation is used. If the effects of mutant genes on the trait are symmetrically distributed and they are unlinked, the variance of mutant effects is not an important parameter. If the distribution is skewed, unless effects or the population size is small, the proportion of mutants that have increasing effect is the critical parameter. With linkage the distribution of genotypic values in the population becomes skewed downward and the equilibrium genetic variance and response are smaller as disequilibrium becomes important. Linkage effects are greater when the mutational variance is contributed by many genes of small effect than few of large effect, and are greater when the majority of mutants increase rather than decrease the trait because genes that are of large effect or are deleterious do not segregate for long. The most likely conditions for ‘‘Muller’s ratchet” are investi- gated. N recent years there has been much interest in the is attained more quickly in the presence of selection I production and maintenance of variation in pop- than in its absence, and is highly dependent on popu- ulations by mutation, stimulated by the presence of lation size. -

Epigenome-Wide Study Identified Methylation Sites Associated With
nutrients Article Epigenome-Wide Study Identified Methylation Sites Associated with the Risk of Obesity Majid Nikpay 1,*, Sepehr Ravati 2, Robert Dent 3 and Ruth McPherson 1,4,* 1 Ruddy Canadian Cardiovascular Genetics Centre, University of Ottawa Heart Institute, 40 Ruskin St–H4208, Ottawa, ON K1Y 4W7, Canada 2 Plastenor Technologies Company, Montreal, QC H2P 2G4, Canada; [email protected] 3 Department of Medicine, Division of Endocrinology, University of Ottawa, the Ottawa Hospital, Ottawa, ON K1Y 4E9, Canada; [email protected] 4 Atherogenomics Laboratory, University of Ottawa Heart Institute, Ottawa, ON K1Y 4W7, Canada * Correspondence: [email protected] (M.N.); [email protected] (R.M.) Abstract: Here, we performed a genome-wide search for methylation sites that contribute to the risk of obesity. We integrated methylation quantitative trait locus (mQTL) data with BMI GWAS information through a SNP-based multiomics approach to identify genomic regions where mQTLs for a methylation site co-localize with obesity risk SNPs. We then tested whether the identified site contributed to BMI through Mendelian randomization. We identified multiple methylation sites causally contributing to the risk of obesity. We validated these findings through a replication stage. By integrating expression quantitative trait locus (eQTL) data, we noted that lower methylation at cg21178254 site upstream of CCNL1 contributes to obesity by increasing the expression of this gene. Higher methylation at cg02814054 increases the risk of obesity by lowering the expression of MAST3, whereas lower methylation at cg06028605 contributes to obesity by decreasing the expression of SLC5A11. Finally, we noted that rare variants within 2p23.3 impact obesity by making the cg01884057 Citation: Nikpay, M.; Ravati, S.; Dent, R.; McPherson, R. -

Selection Response in Finite Populations
Copyright 0 1996 by the Genetics Society of America Selection Responsein Finite Populations Ming Wei, Armando Caballero and William G. Hill Institute of Cell, Animal and Population Biology, University of Edinburgh, King’s Buildings, Edinburgh EH9 3JT, United Kingdom Manuscript received November 28, 1995 Accepted for publication September 5, 1996 ABSTRACT Formulae were derived to predict genetic response under various selection schemes assuming an infinitesimal model. Accountwas taken of genetic drift, gametic (linkage) disequilibrium (Bulmereffect), inbreeding depression, common environmental variance, and both initial segregating variance within families (a;wo)and mutational (&) variance. The cumulative response to selection until generation t(C8) can be approximated as where N, is the effective population size, = Ne& is the genetic variancewithin families at the steady state (or one-half the genic variance, which is unaffected by selection), and D is the inbreeding depression per unit of inbreeding. & is the selection response at generation 0 assuming preselection so that the linkage disequilibrium effect has stabilized. P is the derivative of the logarithm of the asymptotic response with respect to the logarithm of the within-family genetic variance, ie., their relative rate of change. & is the major determinant of the short term selection response, but aL, Ne and p are also important for the long term. A selection method of high accuracy using family information gives a small N, and will lead to a larger- response in the short term and a smaller response in the long term, utilizing mutation less efficiently. ENETIC response to one cycle of selection for a 1975) using all information available are formally the G quantitative character is solely a function of selec- methods to achieve the maximum response for one tion accuracy (the correlation between the selection generation of selection. -

The Qtn Program and the Alleles That Matter for Evolution: All That’S Gold Does Not Glitter
PERSPECTIVE doi:10.1111/j.1558-5646.2011.01486.x THE QTN PROGRAM AND THE ALLELES THAT MATTER FOR EVOLUTION: ALL THAT’S GOLD DOES NOT GLITTER Matthew V. Rockman1,2 1Department of Biology and Center for Genomics and Systems Biology, New York University, 12 Waverly Place, New York, NY 10003 2E-mail: [email protected] Received April 20, 2011 Accepted September 30, 2011 The search for the alleles that matter, the quantitative trait nucleotides (QTNs) that underlie heritable variation within populations and divergence among them, is a popular pursuit. But what is the question to which QTNs are the answer? Although their pursuit is often invoked as a means of addressing the molecular basis of phenotypic evolution or of estimating the roles of evolutionary forces, the QTNs that are accessible to experimentalists, QTNs of relatively large effect, may be uninformative about these issues if large-effect variants are unrepresentative of the alleles that matter. Although 20th century evolutionary biology generally viewed large-effect variants as atypical, the field has recently undergone a quiet realignment toward a view of readily discoverable large- effect alleles as the primary molecular substrates for evolution. I argue that neither theory nor data justify this realignment. Models and experimental findings covering broad swaths of evolutionary phenomena suggest that evolution often acts via large numbers of small-effect polygenes, individually undetectable. Moreover, these small-effect variants are different in kind, at the molecular level, from the large-effect alleles accessible to experimentalists. Although discoverable QTNs address some fundamental evolutionary questions, they are essentially misleading about many others. -

QUANTITATIVE TRAIT LOCUS ANALYSIS: in Several Variants (I.E., Alleles)
TECHNOLOGIES FROM THE FIELD QUANTITATIVE TRAIT LOCUS ANALYSIS: in several variants (i.e., alleles). QTL analysis allows one MULTIPLE CROSS AND HETEROGENEOUS to map, with some precision, the genomic position of STOCK MAPPING these alleles. For many researchers in the alcohol field, the break Robert Hitzemann, Ph.D.; John K. Belknap, Ph.D.; through with respect to the genetic determination of alcohol- and Shannon K. McWeeney, Ph.D. related behaviors occurred when Plomin and colleagues (1991) made the seminal observation that a specific panel of RI mice (i.e., the BXD panel) could be used to identify KEY WORDS: Genetic theory of alcohol and other drug use; genetic the physical location of (i.e., to map) QTLs for behavioral factors; environmental factors; behavioral phenotype; behavioral phenotypes. Because the phenotypes of the different strains trait; quantitative trait gene (QTG); inbred animal strains; in this panel had been determined for many alcohol- recombinant inbred (RI) mouse strains; quantitative traits; related traits, researchers could readily apply the strategy quantitative trait locus (QTL) mapping; multiple cross mapping of RI–QTL mapping (Gora-Maslak et al. 1991). Although (MCM); heterogeneous stock (HS) mapping; microsatellite investigators recognized early on that this panel was not mapping; animal models extensive enough to answer all questions, the emerging data illustrated the rich genetic complexity of alcohol- related phenotypes (Belknap 1992; Plomin and McClearn ntil well into the 1990s, both preclinical and clinical 1993). research focused on finding “the” gene for human The next advance came with the development of diseases, including alcoholism. This focus was rein U microsatellite maps (Dietrich et al. -

HUMAN GENE MAPPING WORKSHOPS C.1973–C.1991
HUMAN GENE MAPPING WORKSHOPS c.1973–c.1991 The transcript of a Witness Seminar held by the History of Modern Biomedicine Research Group, Queen Mary University of London, on 25 March 2014 Edited by E M Jones and E M Tansey Volume 54 2015 ©The Trustee of the Wellcome Trust, London, 2015 First published by Queen Mary University of London, 2015 The History of Modern Biomedicine Research Group is funded by the Wellcome Trust, which is a registered charity, no. 210183. ISBN 978 1 91019 5031 All volumes are freely available online at www.histmodbiomed.org Please cite as: Jones E M, Tansey E M. (eds) (2015) Human Gene Mapping Workshops c.1973–c.1991. Wellcome Witnesses to Contemporary Medicine, vol. 54. London: Queen Mary University of London. CONTENTS What is a Witness Seminar? v Acknowledgements E M Tansey and E M Jones vii Illustrations and credits ix Abbreviations and ancillary guides xi Introduction Professor Peter Goodfellow xiii Transcript Edited by E M Jones and E M Tansey 1 Appendix 1 Photographs of participants at HGM1, Yale; ‘New Haven Conference 1973: First International Workshop on Human Gene Mapping’ 90 Appendix 2 Photograph of (EMBO) workshop on ‘Cell Hybridization and Somatic Cell Genetics’, 1973 96 Biographical notes 99 References 109 Index 129 Witness Seminars: Meetings and publications 141 WHAT IS A WITNESS SEMINAR? The Witness Seminar is a specialized form of oral history, where several individuals associated with a particular set of circumstances or events are invited to meet together to discuss, debate, and agree or disagree about their memories. The meeting is recorded, transcribed, and edited for publication. -

LD5655.V855 1993.R857.Pdf
INITIATING INTERNATIONAL COLLABORATION: A STUDY OF THE HUMAN GENOME ORGANIZATION by Deborah Rumrill Thesis submitted to the faculty of the Virginia Polytechnic Institute and State University in partial fulfillment of the requirements for the degree of MASTER OF SCIENCE in Science and Technology Studies APPROVED: Mj Lallon Doris Zallen Av)eM rg Ann La Berg James Bohland July 1993 Blacksburg, Virginia INITIATING INTERNATIONAL COLLABORATION: A STUDY OF THE HUMAN GENOME ORGANIZATION by Deborah G. C. Rumrill Committee Chair: Doris T. Zallen Department of Science and Technology Studies (ABSTRACT) The formation of the Human Genome Organization, nicknamed HUGO, in 1988 was a response by scientists to the increasing number of programs designed to examine in detail human genetic material that were developing worldwide in the mid 1980s and the perceived need for initiating international collaboration among the genomic researchers. Despite the expectations of its founders, the Human Genome Organization has not attained immediate acceptance either inside or outside the scientific community, struggling since its inception to gain credibility. Although the organization has been successful as well as unsuccessful in its efforts to initiate international collaboration, there has been little or no analysis of the underlying reasons for these outcomes. This study examines the collaborative activities of the organization, which are new to the biological community in terms of kind and scale, and finds two conditions to be influential in the outcome of the organization’s efforts: 1) the prior existence of a model for the type of collaboration attempted; and 2) the existence or creation of a financial or political incentive to accept a new collaborative activity. -

Article the Power to Detect Quantitative Trait Loci Using
The Power to Detect Quantitative Trait Loci Using Resequenced, Experimentally Evolved Populations of Diploid, Sexual Organisms James G. Baldwin-Brown,*,1 Anthony D. Long,1 and Kevin R. Thornton1 1Department of Ecology and Evolutionary Biology, University of California, Irvine *Corresponding author: E-mail: [email protected]. Associate editor: John Parsch Abstract A novel approach for dissecting complex traits is to experimentally evolve laboratory populations under a controlled environment shift, resequence the resulting populations, and identify single nucleotide polymorphisms (SNPs) and/or genomic regions highly diverged in allele frequency. To better understand the power and localization ability of such an evolve and resequence (E&R) approach, we carried out forward-in-time population genetics simulations of 1 Mb genomic regions under a large combination of experimental conditions, then attempted to detect significantly diverged SNPs. Our analysis indicates that the ability to detect differentiation between populations is primarily affected by selection coef- ficient, population size, number of replicate populations, and number of founding haplotypes. We estimate that E&R studies can detect and localize causative sites with 80% success or greater when the number of founder haplotypes is over 500, experimental populations are replicated at least 25-fold, population size is at least 1,000 diploid individuals, and the selection coefficient on the locus of interest is at least 0.1. More achievable experimental designs (less replicated, fewer founder haplotypes, smaller effective population size, and smaller selection coefficients) can have power of greater than 50% to identify a handful of SNPs of which one is likely causative. Similarly, in cases where s 0.2, less demanding experimental designs can yield high power.