Epigenome-Wide Study Identified Methylation Sites Associated With
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Lineage-Specific Mapping of Quantitative Trait Loci
Heredity (2013) 111, 106–113 & 2013 Macmillan Publishers Limited All rights reserved 0018-067X/13 www.nature.com/hdy ORIGINAL ARTICLE Lineage-specific mapping of quantitative trait loci C Chen1 and K Ritland We present an approach for quantitative trait locus (QTL) mapping, termed as ‘lineage-specific QTL mapping’, for inferring allelic changes of QTL evolution along with branches in a phylogeny. We describe and analyze the simplest case: by adding a third taxon into the normal procedure of QTL mapping between pairs of taxa, such inferences can be made along lineages to a presumed common ancestor. Although comparisons of QTL maps among species can identify homology of QTLs by apparent co- location, lineage-specific mapping of QTL can classify homology into (1) orthology (shared origin of QTL) versus (2) paralogy (independent origin of QTL within resolution of map distance). In this light, we present a graphical method that identifies six modes of QTL evolution in a three taxon comparison. We then apply our model to map lineage-specific QTLs for inbreeding among three taxa of yellow monkey-flower: Mimulus guttatus and two inbreeders M. platycalyx and M. micranthus, but critically assuming outcrossing was the ancestral state. The two most common modes of homology across traits were orthologous (shared ancestry of mutation for QTL alleles). The outbreeder M. guttatus had the fewest lineage-specific QTL, in accordance with the presumed ancestry of outbreeding. Extensions of lineage-specific QTL mapping to other types of data and crosses, and to inference of ancestral QTL state, are discussed. Heredity (2013) 111, 106–113; doi:10.1038/hdy.2013.24; published online 24 April 2013 Keywords: Quantitative trait locus mapping; evolution of mating systems; Mimulus INTRODUCTION common ancestor of these two taxa, back to the common ancestor of Knowledge of the genetic architecture of complex traits can offer all three taxa and forwards again to the third taxon. -
Pleiotropic Scaling and QTL Data Arising From: G
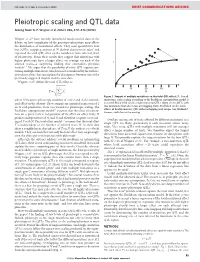
NATURE | Vol 456 | 4 December 2008 BRIEF COMMUNICATIONS ARISING Pleiotropic scaling and QTL data Arising from: G. P. Wagner et al. Nature 452, 470–473 (2008) Wagner et al.1 have recently introduced much-needed data to the debate on how complexity of the genotype–phenotype map affects the distribution of mutational effects. They used quantitative trait loci (QTLs) mapping analysis of 70 skeletal characters in mice2 and regressed the total QTL effect on the number of traits affected (level of pleiotropy). From their results they suggest that mutations with higher pleiotropy have a larger effect, on average, on each of the T affected traits—a surprising finding that contradicts previous models3–7. We argue that the possibility of some QTL regions con- taining multiple mutations, which was not considered by the authors, introduces a bias that can explain the discrepancy between one of the previously suggested models and the new data. Wagner et al.1 define the total QTL effect as vffiffiffiffiffiffiffiffiffiffiffiffiffi u uXN 10 20 30 ~t 2 T Ai N i~1 Figure 1 | Impact of multiple mutations on the total QTL effect, T. For all where N measures pleiotropy (number of traits) and Ai the normal- mutations, strict scaling according to the Euclidean superposition model is ized effect on the ith trait. They compare an empirical regression of T assumed. Black filled circles, single-mutation QTLs. Open circles, QTLs with on N with predictions from two models for pleiotropic scaling. The two mutations that affect non-overlapping traits. Red filled circles, total Euclidean superposition model3,4 assumes that the effect of a muta- effects of double-mutant QTL with overlapping trait ranges (see Methods). -

1 ROBERT PLOMIN on Behavioral Genetics Genetic Links Common
Genetic links • Background – Why has educational psychology ben so slow ROBERT PLOMIN to accept genetics? on Behavioral Genetics • Beyond nature vs. nurture – 3 findings from genetic research with far- reaching implications for education: October, 2004 The nature of nurture The abnormal is normal Generalist genes • Molecular Genetics Quantitative genetics: twin and adoption studies Common medical disorders Identical twins Fraternal twins (monozygotic, MZ) (dizygotic, DZ) 1 Twin concordances in TEDS at 7 years Twins Early Development Study (TEDS) for low reading and maths Perceptions of nature/nurture: % parents (N=1340) and teachers Beyond nature vs. nurture: (N=667) indicating that genes are at least as important as environment (Walker & Plomin, Ed. Psych., in press) The nature of nurture • Genetic influence on experience • Genotype-environment correlation – Children’s genetic propensities are correlated with their experiences passive evocative active 2 Beyond nature vs. nurture: Genotype-environment correlation The nature of nurture • Passive • Genotype-environment correlation – e.g., family background (SES) and school – Children’s genetic propensities are correlated with achievement their experiences passive • Evocative evocative – Children evoke experiences for genetic reasons Active • Active • Molecular genetics – Children actively create environments that foster – Find genes associated with learning experiences their genetic propensities Beyond nature vs. nurture: The one-gene one-disability (OGOD) model The abnormal is normal • Common learning disabilities are the quantitative extreme of the same genetic and environmental factors responsible for normal variation in learning abilities. • There are no common learning disabilities, just the extremes of normal variation. 3 OGOD (rare) and QTL (common) disorders The quantitative trait locus (QTL) model Beyond nature vs. -

CCNL1 (NM 020307) Human Untagged Clone – SC113160 | Origene
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for SC113160 CCNL1 (NM_020307) Human Untagged Clone Product data: Product Type: Expression Plasmids Product Name: CCNL1 (NM_020307) Human Untagged Clone Tag: Tag Free Symbol: CCNL1 Synonyms: ania-6a; ANIA6A; BM-001; PRO1073 Vector: pCMV6-XL5 E. coli Selection: Ampicillin (100 ug/mL) Cell Selection: None Fully Sequenced ORF: >NCBI ORF sequence for NM_020307, the custom clone sequence may differ by one or more nucleotides ATGGCGTCCGGGCCTCATTCGACAGCTACTGCTGCCGCAGCCGCCTCATCGGCCGCCCCAAGCGCGGGCG GCTCCAGCTCCGGGACGACGACCACGACGACGACCACGACGGGAGGGATCCTGATCGGCGATCGCCTGTA CTCGGAAGTTTCACTTACCATCGACCACTCTCTGATTCCGGAGGAGAGGCTCTCGCCCACCCCATCCATG CAGGATGGGCTCGACCTGCCCAGTGAGACGGACTTACGCATCCTGGGCTGCGAGCTCATCCAGGCCGCCG GCATTCTCCTCCGGCTGCCGCAGGTGGCGATGGCAACGGGGCAGGTGTTGTTTCATCGTTTTTTCTACTC CAAATCTTTCGTCAAACACAGTTTCGAGATTGTTGCTATGGCTTGTATTAATCTTGCATCAAAAATCGAA GAAGCACCTAGAAGAATAAGAGATGTGATTAATGTATTCCACCACCTCCGCCAGTTAAGAGGAAAAAGGA CTCCAAGCCCCCTGATCCTTGATCAGAACTACATTAACACCAAAAATCAAGTTATCAAAGCAGAGAGGAG GGTGCTAAAGGAGTTGGGATTTTGTGTTCATGTCAAGCATCCTCATAAGATCATTGTTATGTATTTACAA GTCTTAGAATGTGAACGTAATCAAACCCTGGTTCAAACTGCCTGGAATTACATGAATGACAGTCTTCGAA CCAATGTGTTTGTTCGATTTCAACCAGAGACTATAGCATGTGCTTGCATCTACCTTGCAGCTAGAGCACT TCAGATTCCGTTGCCAACTCGTCCCCATTGGTTTCTTCTTTTTGGTACTACAGAAGAGGAAATCCAGGAA ATCTGCATAGAAACACTTAGGCTTTATACCAGAAAAAAGCCAAACTATGAATTACTGGAAAAAGAAGTAG AAAAAAGAAAAGTAGCCTTACAAGAAGCCAAATTAAAAGCAAAGGGATTGAATCCGGATGGAACTCCAGC -

QUANTITATIVE TRAIT LOCUS ANALYSIS: in Several Variants (I.E., Alleles)
TECHNOLOGIES FROM THE FIELD QUANTITATIVE TRAIT LOCUS ANALYSIS: in several variants (i.e., alleles). QTL analysis allows one MULTIPLE CROSS AND HETEROGENEOUS to map, with some precision, the genomic position of STOCK MAPPING these alleles. For many researchers in the alcohol field, the break Robert Hitzemann, Ph.D.; John K. Belknap, Ph.D.; through with respect to the genetic determination of alcohol- and Shannon K. McWeeney, Ph.D. related behaviors occurred when Plomin and colleagues (1991) made the seminal observation that a specific panel of RI mice (i.e., the BXD panel) could be used to identify KEY WORDS: Genetic theory of alcohol and other drug use; genetic the physical location of (i.e., to map) QTLs for behavioral factors; environmental factors; behavioral phenotype; behavioral phenotypes. Because the phenotypes of the different strains trait; quantitative trait gene (QTG); inbred animal strains; in this panel had been determined for many alcohol- recombinant inbred (RI) mouse strains; quantitative traits; related traits, researchers could readily apply the strategy quantitative trait locus (QTL) mapping; multiple cross mapping of RI–QTL mapping (Gora-Maslak et al. 1991). Although (MCM); heterogeneous stock (HS) mapping; microsatellite investigators recognized early on that this panel was not mapping; animal models extensive enough to answer all questions, the emerging data illustrated the rich genetic complexity of alcohol- related phenotypes (Belknap 1992; Plomin and McClearn ntil well into the 1990s, both preclinical and clinical 1993). research focused on finding “the” gene for human The next advance came with the development of diseases, including alcoholism. This focus was rein U microsatellite maps (Dietrich et al. -

Supplementary Materials
Supplementary materials Supplementary Table S1: MGNC compound library Ingredien Molecule Caco- Mol ID MW AlogP OB (%) BBB DL FASA- HL t Name Name 2 shengdi MOL012254 campesterol 400.8 7.63 37.58 1.34 0.98 0.7 0.21 20.2 shengdi MOL000519 coniferin 314.4 3.16 31.11 0.42 -0.2 0.3 0.27 74.6 beta- shengdi MOL000359 414.8 8.08 36.91 1.32 0.99 0.8 0.23 20.2 sitosterol pachymic shengdi MOL000289 528.9 6.54 33.63 0.1 -0.6 0.8 0 9.27 acid Poricoic acid shengdi MOL000291 484.7 5.64 30.52 -0.08 -0.9 0.8 0 8.67 B Chrysanthem shengdi MOL004492 585 8.24 38.72 0.51 -1 0.6 0.3 17.5 axanthin 20- shengdi MOL011455 Hexadecano 418.6 1.91 32.7 -0.24 -0.4 0.7 0.29 104 ylingenol huanglian MOL001454 berberine 336.4 3.45 36.86 1.24 0.57 0.8 0.19 6.57 huanglian MOL013352 Obacunone 454.6 2.68 43.29 0.01 -0.4 0.8 0.31 -13 huanglian MOL002894 berberrubine 322.4 3.2 35.74 1.07 0.17 0.7 0.24 6.46 huanglian MOL002897 epiberberine 336.4 3.45 43.09 1.17 0.4 0.8 0.19 6.1 huanglian MOL002903 (R)-Canadine 339.4 3.4 55.37 1.04 0.57 0.8 0.2 6.41 huanglian MOL002904 Berlambine 351.4 2.49 36.68 0.97 0.17 0.8 0.28 7.33 Corchorosid huanglian MOL002907 404.6 1.34 105 -0.91 -1.3 0.8 0.29 6.68 e A_qt Magnogrand huanglian MOL000622 266.4 1.18 63.71 0.02 -0.2 0.2 0.3 3.17 iolide huanglian MOL000762 Palmidin A 510.5 4.52 35.36 -0.38 -1.5 0.7 0.39 33.2 huanglian MOL000785 palmatine 352.4 3.65 64.6 1.33 0.37 0.7 0.13 2.25 huanglian MOL000098 quercetin 302.3 1.5 46.43 0.05 -0.8 0.3 0.38 14.4 huanglian MOL001458 coptisine 320.3 3.25 30.67 1.21 0.32 0.9 0.26 9.33 huanglian MOL002668 Worenine -

High-Throughput Bioinformatics Approaches to Understand Gene Expression Regulation in Head and Neck Tumors
High-throughput bioinformatics approaches to understand gene expression regulation in head and neck tumors by Yanxiao Zhang A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Bioinformatics) in The University of Michigan 2016 Doctoral Committee: Associate Professor Maureen A. Sartor, Chair Professor Thomas E. Carey Assistant Professor Hui Jiang Professor Ronald J. Koenig Associate Professor Laura M. Rozek Professor Kerby A. Shedden c Yanxiao Zhang 2016 All Rights Reserved I dedicate this thesis to my family. For their unfailing love, understanding and support. ii ACKNOWLEDGEMENTS I would like to express my gratitude to Dr. Maureen Sartor for her guidance in my research and career development. She is a great mentor. She patiently taught me when I started new in this field, granted me freedom to explore and helped me out when I got lost. Her dedication to work, enthusiasm in teaching, mentoring and communicating science have inspired me to feel the excite- ment of research beyond novel scientific discoveries. I’m also grateful to have an interdisciplinary committee. Their feedback on my research progress and presentation skills is very valuable. In particular, I would like to thank Dr. Thomas Carey and Dr. Laura Rozek for insightful discussions on the biology of head and neck cancers and human papillomavirus, Dr. Ronald Koenig for expert knowledge on thyroid cancers, Dr. Hui Jiang and Dr. Kerby Shedden for feedback on the statistics part of my thesis. I would like to thank all the past and current members of Sartor lab for making the lab such a lovely place to stay and work in. -

Article the Power to Detect Quantitative Trait Loci Using
The Power to Detect Quantitative Trait Loci Using Resequenced, Experimentally Evolved Populations of Diploid, Sexual Organisms James G. Baldwin-Brown,*,1 Anthony D. Long,1 and Kevin R. Thornton1 1Department of Ecology and Evolutionary Biology, University of California, Irvine *Corresponding author: E-mail: [email protected]. Associate editor: John Parsch Abstract A novel approach for dissecting complex traits is to experimentally evolve laboratory populations under a controlled environment shift, resequence the resulting populations, and identify single nucleotide polymorphisms (SNPs) and/or genomic regions highly diverged in allele frequency. To better understand the power and localization ability of such an evolve and resequence (E&R) approach, we carried out forward-in-time population genetics simulations of 1 Mb genomic regions under a large combination of experimental conditions, then attempted to detect significantly diverged SNPs. Our analysis indicates that the ability to detect differentiation between populations is primarily affected by selection coef- ficient, population size, number of replicate populations, and number of founding haplotypes. We estimate that E&R studies can detect and localize causative sites with 80% success or greater when the number of founder haplotypes is over 500, experimental populations are replicated at least 25-fold, population size is at least 1,000 diploid individuals, and the selection coefficient on the locus of interest is at least 0.1. More achievable experimental designs (less replicated, fewer founder haplotypes, smaller effective population size, and smaller selection coefficients) can have power of greater than 50% to identify a handful of SNPs of which one is likely causative. Similarly, in cases where s 0.2, less demanding experimental designs can yield high power. -

Glossary/Index
Glossary 03/08/2004 9:58 AM Page 119 GLOSSARY/INDEX The numbers after each term represent the chapter in which it first appears. additive 2 allele 2 When an allele’s contribution to the variation in a One of two or more alternative forms of a gene; a single phenotype is separately measurable; the independent allele for each gene is inherited separately from each effects of alleles “add up.” Antonym of nonadditive. parent. ADHD/ADD 6 Alzheimer’s disease 5 Attention Deficit Hyperactivity Disorder/Attention A medical disorder causing the loss of memory, rea- Deficit Disorder. Neurobehavioral disorders character- soning, and language abilities. Protein residues called ized by an attention span or ability to concentrate that is plaques and tangles build up and interfere with brain less than expected for a person's age. With ADHD, there function. This disorder usually first appears in persons also is age-inappropriate hyperactivity, impulsive over age sixty-five. Compare to early-onset Alzheimer’s. behavior or lack of inhibition. There are several types of ADHD: a predominantly inattentive subtype, a predomi- amino acids 2 nantly hyperactive-impulsive subtype, and a combined Molecules that are combined to form proteins. subtype. The condition can be cognitive alone or both The sequence of amino acids in a protein, and hence pro- cognitive and behavioral. tein function, is determined by the genetic code. adoption study 4 amnesia 5 A type of research focused on families that include one Loss of memory, temporary or permanent, that can result or more children raised by persons other than their from brain injury, illness, or trauma. -

Combinatorial Strategies Using CRISPR/Cas9 for Gene Mutagenesis in Adult Mice
Combinatorial strategies using CRISPR/Cas9 for gene mutagenesis in adult mice Avery C. Hunker A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Washington 2019 Reading Committee: Larry S. Zweifel, Chair Sheri J. Mizumori G. Stanley McKnight Program Authorized to Offer Degree: Pharmacology 2 © Copyright 2019 Avery C. Hunker 3 University of Washington ABSTRACT Combinatorial strategies using CRISPR/Cas9 for gene mutagenesis in adult mice Avery C. Hunker Chair of the Supervisory Committee: Larry Zweifel Department of Pharmacology A major challenge to understanding how genes modulate complex behaviors is the inability to restrict genetic manipulations to defined cell populations or circuits. To circumvent this, we created a simple strategy for limiting gene knockout to specific cell populations using a viral-mediated, conditional CRISPR/SaCas9 system in combination with intersectional genetic strategies. A small single guide RNA (sgRNA) directs Staphylococcus aureus CRISPR-associated protein (SaCas9) to unique sites on DNA in a Cre-dependent manner resulting in double strand breaks and gene mutagenesis in vivo. To validate this technique we targeted nine different genes of diverse function in distinct cell types in mice and performed an array of analyses to confirm gene mutagenesis and subsequent protein loss, including IHC, cell-type specific DNA sequencing, electrophysiology, Western blots, and behavior. We show that these vectors are as efficient as conventional conditional gene knockout and provide a viable alternative to complex genetic crosses. This strategy provides additional benefits of 4 targeting gene mutagenesis to cell types previously difficult to isolate, and the ability to target genes in specific neural projections for gene inactivation. -

Mapping Quantitative Trait Loci in Selected Breeding Populations: a Segregation Distortion Approach
Heredity (2015) 115, 538–546 & 2015 Macmillan Publishers Limited All rights reserved 0018-067X/15 www.nature.com/hdy ORIGINAL ARTICLE Mapping quantitative trait loci in selected breeding populations: A segregation distortion approach Y Cui1,2, F Zhang1,JXu1,3,ZLi1 and S Xu2 Quantitative trait locus (QTL) mapping is often conducted in line-crossing experiments where a sample of individuals is randomly selected from a pool of all potential progeny. QTLs detected from such an experiment are important for us to understand the genetic mechanisms governing a complex trait, but may not be directly relevant to plant breeding if they are not detected from the breeding population where selection is targeting for. QTLs segregating in one population may not necessarily segregate in another population. To facilitate marker-assisted selection, QTLs must be detected from the very population which the selection is targeting. However, selected breeding populations often have depleted genetic variation with small population sizes, resulting in low power in detecting useful QTLs. On the other hand, if selection is effective, loci controlling the selected trait will deviate from the expected Mendelian segregation ratio. In this study, we proposed to detect QTLs in selected breeding populations via the detection of marker segregation distortion in either a single population or multiple populations using the same selection scheme. Simulation studies showed that QTL can be detected in strong selected populations with selected population sizes as small as 25 plants. We applied the new method to detect QTLs in two breeding populations of rice selected for high grain yield. Seven QTLs were identified, four of which have been validated in advanced generations in a follow-up study. -

Pleiotropic Patterns of Quantitative Trait Loci for 70 Murine Skeletal Traits
Copyright Ó 2008 by the Genetics Society of America DOI: 10.1534/genetics.107.084434 Pleiotropic Patterns of Quantitative Trait Loci for 70 Murine Skeletal Traits Jane P. Kenney-Hunt,*,1 Bing Wang,* Elizabeth A. Norgard,* Gloria Fawcett,* Doug Falk,* L. Susan Pletscher,* Joseph P. Jarvis,* Charles Roseman,† Jason Wolf‡ and James M. Cheverud* *Department of Anatomy and Neurobiology, Washington University School of Medicine, St. Louis, Missouri 63110, †Department of Anthropology, University of Illinois, Urbana, Illinois 61801 and ‡Faculty of Life Sciences, University of Manchester, Manchester M13 9PT, United Kingdom Manuscript received November 11, 2007 Accepted for publication February 1, 2008 ABSTRACT Quantitative trait locus (QTL) studies of a skeletal trait or a few related skeletal components are becoming commonplace, but as yet there has been no investigation of pleiotropic patterns throughout the skeleton. We present a comprehensive survey of pleiotropic patterns affecting mouse skeletal morphology in an intercross of LG/J and SM/J inbred strains (N ¼ 1040), using QTL analysis on 70 skeletal traits. We identify 798 single- trait QTL, coalescing to 105 loci that affect on average 7–8 traits each. The number of traits affected per locus ranges from only 1 trait to 30 traits. Individual traits average 11 QTL each, ranging from 4 to 20. Skeletal traits are affected by many, small-effect loci. Significant additive genotypic values average 0.23 standard deviation (SD) units. Fifty percent of loci show codominance with heterozygotes having intermediate phenotypic values. When dominance does occur, the LG/J allele tends to be dominant to the SM/J allele (30% vs.