Assessment Report
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Hepatitis B Vaccine Is the Most Widely Used Vaccine in the World, with Over 1 Billion Doses Given
Hepatitis B Vaccine Protect Yourself and Those You Love What is Hepatitis B? Hepatitis B is the most common serious liver infection in the world. It is caused by the hepatitis B virus (HBV), which attacks liver cells and can lead to liver failure, cirrhosis (scarring), or liver cancer later in life. The virus is transmitted through direct contact with infected blood and bodily fluids, and from an infected woman to her newborn at birth. Is there a safe vaccine for hepatitis B? YES! The good news is that there is a safe and effective vaccine for hepatitis B. The vaccine is a series, typically given as three shots over a six-month period that will provide a lifetime of protection. You cannot get hepatitis B from the vaccine – there is no human blood or live virus in the vaccine. The hepatitis B vaccine is the most widely used vaccine in the world, with over 1 billion doses given. The hepatitis B vaccine is the first "anti-cancer" vaccine because it can help prevent liver cancer! Who should be vaccinated against hepatitis B? The World Health Organization (WHO) and the U.S. Centers for Disease Control and Prevention (CDC) recommend the hepatitis B vaccine for all newborns and children up to 18 years of age, and all high-risk adults. All infants should receive the first dose of the vaccine in the delivery room or in the first 24 hours of life, preferably within 12 hours. (CDC recommends the first dose within 12 hours vs. the WHO recommendation of 24 hours.) The HBV vaccine is recommended to anyone who is at high risk of infection. -

Engerix-B Data Sheet
NEW ZEALAND DATA SHEET 1. PRODUCT NAME ENGERIX-B 20 micrograms/mL, suspension for injection. ENGERIX-B paediatric 10 micrograms/0.5 mL, suspension for injection. 2. QUALITATIVE AND QUANTITATIVE COMPOSITION ENGERIX-B paediatric dose: 10 microgram (µg) dose vaccine 1 dose (0.5 mL) contains: Hepatitis B surface antigen 1, 2 10 micrograms 1Adsorbed on aluminium hydroxide, hydrated Total: 0.25 milligrams Al3+ 2Produced in yeast cells (Saccharomyces cerevisiae) by recombinant DNA technology ENGERIX-B: 20 microgram (µg) dose vaccine 1 dose (1 mL) contains: Hepatitis B surface antigen1, 2 20 micrograms 1Adsorbed on aluminium hydroxide, hydrated Total: 0.50 milligrams Al3+ 2Produced in yeast cells (Saccharomyces cerevisiae) by recombinant DNA technology The vaccine is highly purified, and meets the WHO requirements for recombinant hepatitis B vaccines. No substances of human origin are used in its manufacture. For the full list of excipients, see section 6.1 List of excipients. 3. PHARMACEUTICAL FORM Suspension for injection. ENGERIX-B is a turbid white suspension. Upon storage, a fine white deposit with a clear colourless supernatant may be observed. 4. CLINICAL PARTICULARS 4.1 Therapeutic indications ENGERIX-B is indicated for active immunisation against hepatitis B virus infection. As part of the national immunisation schedule, the New Zealand Ministry of Health* recommend all infants, unvaccinated children up to the age of 16 years, and household and sexual contacts of known hepatitis B carriers receive a primary course of vaccination against hepatitis B. 1 Immunisation is also recommended for seronegative persons who are at substantial risk and have been demonstrated or judged to be susceptible to the hepatitis B virus (HBV). -

Hepatitis B Vaccine – Frequently Asked Questions (Information from the CDC)
AAMC Standardized Immunization Form 2020 Hepatitis B Vaccine – Frequently Asked Questions (Information from the CDC) 1. What are the hepatitis B vaccines licensed for use in the United States? Three single-antigen vaccines and two combination vaccines are currently licensed in the United States. Single-antigen hepatitis B vaccines: • ENGERIX-B® • RECOMBIVAX HB® • HEPLISAV-B™ Combination vaccines: • PEDIARIX®: Combined hepatitis B, diphtheria, tetanus, acellular pertussis (DTaP), and inactivated poliovirus (IPV) vaccine. Cannot be administered before age 6 weeks or after age 7 years. • TWINRIX®: Combined Hepatitis A and hepatitis B vaccine. Recommended for people aged ≥18 years who are at increased risk for both HAV and HBV infections. 2. What are the recommended schedules for hepatitis B vaccination? The vaccination schedule most often used for children and adults is three doses given at 0, 1, and 6 months. Alternate schedules have been approved for certain vaccines and/or populations. A new formulation, Heplisav-B (HepB-CpG), is approved to be given as two doses one month apart. 3. If there is an interruption between doses of hepatitis B vaccine, does the vaccine series need to be restarted? No. The series does not need to be restarted but the following should be considered: • If the vaccine series was interrupted after the first dose, the second dose should be administered as soon as possible. • The second and third doses should be separated by an interval of at least 8 weeks. • If only the third dose is delayed, it should be administered as soon as possible. 4. Is it harmful to administer an extra dose of hepatitis B vaccine or to repeat the entire vaccine series if documentation of the vaccination history is unavailable or the serology test is negative? No, administering extra doses of single-antigen hepatitis B vaccine is not harmful. -

Recommended Adult Immunization Schedule
Recommended Adult Immunization Schedule UNITED STATES for ages 19 years or older 2021 Recommended by the Advisory Committee on Immunization Practices How to use the adult immunization schedule (www.cdc.gov/vaccines/acip) and approved by the Centers for Disease Determine recommended Assess need for additional Review vaccine types, Control and Prevention (www.cdc.gov), American College of Physicians 1 vaccinations by age 2 recommended vaccinations 3 frequencies, and intervals (www.acponline.org), American Academy of Family Physicians (www.aafp. (Table 1) by medical condition and and considerations for org), American College of Obstetricians and Gynecologists (www.acog.org), other indications (Table 2) special situations (Notes) American College of Nurse-Midwives (www.midwife.org), and American Academy of Physician Assistants (www.aapa.org). Vaccines in the Adult Immunization Schedule* Report y Vaccines Abbreviations Trade names Suspected cases of reportable vaccine-preventable diseases or outbreaks to the local or state health department Haemophilus influenzae type b vaccine Hib ActHIB® y Clinically significant postvaccination reactions to the Vaccine Adverse Event Hiberix® Reporting System at www.vaers.hhs.gov or 800-822-7967 PedvaxHIB® Hepatitis A vaccine HepA Havrix® Injury claims Vaqta® All vaccines included in the adult immunization schedule except pneumococcal 23-valent polysaccharide (PPSV23) and zoster (RZV) vaccines are covered by the Hepatitis A and hepatitis B vaccine HepA-HepB Twinrix® Vaccine Injury Compensation Program. Information on how to file a vaccine injury Hepatitis B vaccine HepB Engerix-B® claim is available at www.hrsa.gov/vaccinecompensation. Recombivax HB® Heplisav-B® Questions or comments Contact www.cdc.gov/cdc-info or 800-CDC-INFO (800-232-4636), in English or Human papillomavirus vaccine HPV Gardasil 9® Spanish, 8 a.m.–8 p.m. -

Vaccinations for Adults with Chronic Liver Disease Or Infection
Vaccinations for Adults with Chronic Liver Disease or Infection This table shows which vaccinations you should have to protect your health if you have chronic hepatitis B or C infection or chronic liver disease (e.g., cirrhosis). Make sure you and your healthcare provider keep your vaccinations up to date. Vaccine Do you need it? Hepatitis A Yes! Your chronic liver disease or infection puts you at risk for serious complications if you get infected with the (HepA) hepatitis A virus. If you’ve never been vaccinated against hepatitis A, you need 2 doses of this vaccine, usually spaced 6–18 months apart. Hepatitis B Yes! If you already have chronic hepatitis B infection, you won’t need hepatitis B vaccine. However, if you have (HepB) hepatitis C or other causes of chronic liver disease, you do need hepatitis B vaccine. The vaccine is given in 2 or 3 doses, depending on the brand. Ask your healthcare provider if you need screening blood tests for hepatitis B. Hib (Haemophilus Maybe. Some adults with certain high-risk conditions, for example, lack of a functioning spleen, need vaccination influenzae type b) with Hib. Talk to your healthcare provider to find out if you need this vaccine. Human Yes! You should get this vaccine if you are age 26 years or younger. Adults age 27 through 45 may also be vacci- papillomavirus nated against HPV after a discussion with their healthcare provider. The vaccine is usually given in 3 doses over a (HPV) 6-month period. Influenza Yes! You need a dose every fall (or winter) for your protection and for the protection of others around you. -

Hepatitis B Factsheet: COVID-19 and Hep B
Hepatitis B factsheet: COVID-19 and hep B For more information about anything in this factsheet, phone the Hepatitis Infoline on 1800 803 990 or go to www.hep.org.au COVID-19 and hep B Frequently Asked Questions (FAQ) There’s so much information swirling around these days as we deal with the changing world around us due to COVID-19. There’s lots of incorrect information and it’s easy to be overwhelmed and confused. On this page, we’ll try to clear some things up. It is important that information is helpful, clear, and checked and confirmed by experts. Please share the following information – it comes from health and medical specialists. COVID-19 is an acronym that stands for COronaVIrus Disease 2019. You might hear it called coronavirus, corona, SARS-CoV-2, or even rona. We’ll call it COVID-19 or coronavirus in this FAQ but all these terms are essentially talking about the same thing and we’ll explain them at the bottom. You’ll also hear new or unfamiliar terms like “social distancing,” “physical distancing,” “self-isolation,” and “quarantine”. These terms and what they actually mean can become confusing so we’ve including a glossary at the bottom of this FAQ. This virus has only been known about since around December 2019 so it’s all very new, information changes quickly, and we’re all learning day-by-day and week-by-week. Compare COVID-19 to hep B which we’ve known about since the 1980s and hep B even earlier in the 1960s! We’ll regularly update this page as new information comes to light. -

Hepatitis B Vaccine W H a T Y O U N E E D T O K N O W
HEPATITIS B VACCINE W H A T Y O U N E E D T O K N O W 1 What is hepatitis B? Hepatitis B vaccine: Why get 2 vaccinated? Hepatitis B is a serious disease that affects the liver. It is caused by the hepatitis B virus (HBV). HBV Hepatitis B vaccine can prevent hepatitis B, and can cause: the serious consequences of HBV infection, including liver cancer and cirrhosis. Acute (short-term) illness. This can lead to: • loss of appetite • diarrhea and vomiting Routine hepatitis B vaccination of U.S. children • tiredness • jaundice (yellow skin or eyes) began in 1991. Since then, the reported incidence of • pain in muscles, joints, and stomach acute hepatitis B among children and adolescents has dropped by more than 95% – and by 75% in all Acute illness is more common among adults. age groups. Children who become infected usually do not have acute illness. Hepatitis B vaccine is made from a part of the hepatitis B virus. It cannot cause HBV infection. Chronic (long-term) infection. Some people go on to develop chronic HBV infection. This can be very Hepatitis B vaccine is usually given as a series of 3 serious, and often leads to: or 4 shots. This vaccine series gives long-term •liver damage (cirrhosis) •liver cancer •death protection from HBV infection, possibly lifelong. Chronic infection is more common among infants Who should get hepatitis B and children than among adults. People who are 3 vaccine and when? infected can spread HBV to others, even if they don’t appear sick. -
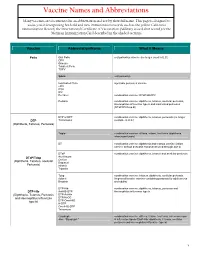
Vaccine Names and Abbreviations Vaccine Names and Abbreviations Many Vaccines Are Documented in an Abbreviation and Not by Their Full Name
Vaccine Names and Abbreviations Vaccine Names and Abbreviations Many vaccines are documented in an abbreviation and not by their full name. This page is designed to assist you in interpreting both old and new immunization records such as the yellow California Immunization Record, the International Certificate of Vaccination (Military issued shot record) or the Mexican Immunization Card described in the shaded sections. Vaccine Abbreviation/Name What it Means Polio Oral Polio oral poliovirus vaccine (no longer used in U.S.) OPV Orimune Trivalent Polio TOPV Sabin oral poliovirus Inactivated Polio injectable poliovirus vaccine eIPV IPOL IPV Pentacel combination vaccine: DTaP/Hib/IPV Pediarix combination vaccine: diphtheria, tetanus, acellular pertussis, Haemophilus influenzae type b and inactivated poliovirus (DTaP/IPV/Hep B) DTP or DPT combination vaccine: diphtheria, tetanus, pertussis (no longer DTP Tri-Immunol available in U.S.) (Diphtheria, Tetanus, Pertussis) Triple combination vaccine: difteria, tétano, tos ferina (diphtheria, tetanus pertussis) DT combination vaccine: diphtheria and tetanus vaccine (infant vaccine without pertussis component used through age 6) DTaP combination vaccine: diphtheria, tetanus and acellular pertussis DTaP/Tdap Acel-Imune Certiva (Diphtheria, Tetanus, acellular Daptacel Pertussis) Infanrix Tripedia Tdap combination vaccine: tetanus, diphtheria, acellular pertussis Adacel (improved booster vaccine containing pertussis for adolescents Boostrix and adults) DTP-Hib combination vaccine: diphtheria, tetanus, -

FAQ: Questions You May Have About COVID-19 Vaccines
FAQ: Questions You May Have about COVID-19 Vaccines 1. I’ve heard on the news that the UK is planning on delaying administration of second doses to increase the number of people getting first doses. Does MA DPH have a plan to do that in Massachusetts? The US FDA has made a strong statement against that approach due to the absence of data to suggest it would be sufficiently effective. At this time, there is no plan for the US or the state to recommend delaying administration of the second dose. 2. Is it expected that the Pfizer vaccine may prevent asymptomatic spread similar to the data that is being shown on the Moderna vaccine? Should those who received Pfizer consider being re-vaccinated? While we are awaiting data, there is good reason to hope that the Pfizer vaccine might prevent spread of the SARS CoV-2 virus as the Moderna vaccine does. The two vaccines are very similar. There are no recommendations to re-vaccinate anyone currently. 3. Are there studies looking at whether vaccinated individuals can have asymptomatic COVID and transmit the disease? Yes, these studies are ongoing 4. What is the current recommendation re: COVID vaccination for 16-17 year olds? Current recommendations are to vaccinate adults and certain adolescents aged 16-17 with underlying health conditions (see CDC ACIP guidance https://www.cdc.gov/vaccines/covid- 19/info-by-product/clinical-considerations.html). Only the Pfizer vaccine is authorized by the FDA for persons aged 16-17. 5. How protected are you if you have only received the initial vaccine dose? Because of the very short time period between maximal immunity from the first dose and receipt of the next dose, the level of immunity that can be expected from one dose alone is not clear. -

Medicare Human Services (DHHS) Centers for Medicare & Carriers Manual Medicaid Services (CMS) Part 3 - Claims Process
Department of Health & Medicare Human Services (DHHS) Centers for Medicare & Carriers Manual Medicaid Services (CMS) Part 3 - Claims Process Transmittal 1778 Date: NOVEMBER 1, 2002 CHANGE REQUEST 2392 HEADER SECTION NUMBERS PAGES TO INSERT PAGES TO DELETE 4480.3 - 4480.4 4-313 – 4-314 (2 pp.) 4-313 – 4-314 (2 pp.) NEW/REVISED MATERIAL--EFFECTIVE DATE: January 1, 2003 IMPLEMENTATION DATE: January 1, 2003 Section 4480.2, HCPCS Coding, is being updated to reflect a few minor subgroup typographical references. The following hepatitis B vaccine codes are no longer valid for Medicare purposes, 90740, 90743, 90744, 90746, and 90747. These codes have been replaced with Q3021, Q3022 and Q3023. It has been determined that vaccine codes 90723 and 90748 were inappropriate for this preventive benefit and have therefore been removed from the manual. These codes that are no longer applicable to Medicare will not have a 90-day grace period. These instructions should be implemented within your current operating budget. DISCLAIMER: The revision date and transmittal number only apply to the redlined material. All other material was previously published in the manual and is only being reprinted. CMS-Pub. 14-3 11-02 CLAIMS REVIEW AND ADJUDICATION PROCEDURES 4480.3 Code Description 90732 Pneumococcal polysaccharide vaccine, 23-valent, adult or immunosuppressed patient dosage, for subcutaneous or intramuscular use; Q3021 Injection, hepatitis B vaccine,pediatric or adolescent, per dose; Q3022 Injection, hepatitis B vaccine, adult, per dose and, Q3023 Injeciton, hepatitis B vaccine, immunosuppressed patients (including renal dialysis patients), per dose. The type of service (TOS) for these new Q codes is 1. -

Hepatitis B Vaccine
Hepatitis B Vaccine Addressing Common Questions about Hepatitis B Vaccination for Adults What disease does this vaccine protect against? Hepatitis B vaccine can protect against hepatitis B virus, and the serious consequences of hepatitis B infection, including liver What could happen if I get cancer, liver damage, and liver failure. this disease? How common is this disease? Hepatitis B can cause acute (short-term) illness that can lead to loss of appetite, In the United States, an estimated 800,000 to 1.4 million persons tiredness, pain in muscles, joints, and have chronic (long-term) hepatitis B virus infection. In 2011, stomach, jaundice, diarrhea, and vomiting. there were an estimated 18,000 new hepatitis B virus infections in the United States. Many people don’t know they are infected • Some people, mostly infants and or may not have symptoms and therefore never seek young children, go on to develop medical treatment. chronic hepatitis B infection once they are infected. How is this disease spread? • While most of the people infected do Hepatitis B virus is easily spread through contact with the blood not have symptoms, the infection is or other body fluids of an infected person. People can also still very serious, and can lead to liver be infected from contact with an object contaminated with damage (cirrhosis), liver cancer, and hepatitis B virus. The virus can live at least 7 days outside of the even death. body. People who are chronically infected can spread hepatitis B virus to others. Who is at risk for this disease? Although anyone can get hepatitis B infection, some adults are at greater risk. -

Uproar Over a Little-Known Preservative, Thimerosal, Jostles U.S
Uproar over a little-known preservative, thimerosal, jostles U.S. hepatitis B vaccination policy In 1997, when Frank Pallone, a Democratic congressman from New Jersey, attached a simple amendment to an FDA reauthorization bill, he could not have predicted that it would cause such a commotion two years later. His amendment ran just 133 words. It gave FDA two years to "compile a list of drugs and foods that contain intentionally introduced mercury compounds and … [to] provide a quantitative and qualitative analysis of the mercury compounds in the list…." The bill later evolved into the landmark FDA Modernization Act of 1997 (FDAMA) and was signed into law on November 21, 1997. Pallone’s amendment undoubtedly sprang from his long interest in environmental causes. But he had unwittingly set into motion a chain of events that would, two years later, bring turmoil to the immunization policy world and fears of harm to the nation’s hepatitis B control effort. Thimerosal, old soldier under a cloud At first glance, someone looking for a controversy would not choose thimerosal. It has been used as a vaccine preservative since the 1930s, and, until recently, it has generally been viewed as a safe, reliable, and somewhat drab defender against bacterial and fungal contamination. The compound garners only one short paragraph in the 1249-page Plotkin and Orenstein reference book, Vaccines 3rd Edition (1999). Thimerosal is sometimes added to vaccines during manufacturing as a guarantee against production-related contamination. Its greatest value, however, is in the field, where it acts as a fail-safe against imperfect aseptic handling.