Assessing the Strengths and Weaknesses of Study Designs and Analytic Methods for Comparative Effectiveness Research
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
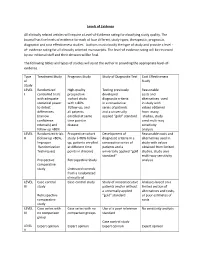
Levels of Evidence Table
Levels of Evidence All clinically related articles will require a Level-of-Evidence rating for classifying study quality. The Journal has five levels of evidence for each of four different study types; therapeutic, prognostic, diagnostic and cost effectiveness studies. Authors must classify the type of study and provide a level - of- evidence rating for all clinically oriented manuscripts. The level-of evidence rating will be reviewed by our editorial staff and their decision will be final. The following tables and types of studies will assist the author in providing the appropriate level-of- evidence. Type Treatment Study Prognosis Study Study of Diagnostic Test Cost Effectiveness of Study Study LEVEL Randomized High-quality Testing previously Reasonable I controlled trials prospective developed costs and with adequate cohort study diagnostic criteria alternatives used statistical power with > 80% in a consecutive in study with to detect follow-up, and series of patients values obtained differences all patients and a universally from many (narrow enrolled at same applied “gold” standard studies, study confidence time point in used multi-way intervals) and disease sensitivity follow up >80% analysis LEVEL Randomized trials Prospective cohort Development of Reasonable costs and II (follow up <80%, study (<80% follow- diagnostic criteria in a alternatives used in Improper up, patients enrolled consecutive series of study with values Randomization at different time patients and a obtained from limited Techniques) points in disease) universally -

Adaptive Clinical Trials: an Introduction
Adaptive clinical trials: an introduction What are the advantages and disadvantages of adaptive clinical trial designs? How and why were Introduction adaptive clinical Adaptive clinical trial design is trials developed? becoming a hot topic in healthcare research, with some researchers In 2004, the FDA published a report arguing that adaptive trials have the on the problems faced by the potential to get new drugs to market scientific community in developing quicker. In this article, we explain new medical treatments.2 The what adaptive trials are and why they report highlighted that the pace of were developed, and we explore both innovation in biomedical science is the advantages of adaptive designs outstripping the rate of advances and the concerns being raised by in the available technologies and some in the healthcare community. tools for evaluating new treatments. Outdated tools are being used to assess new treatments and there What are adaptive is a critical need to improve the effectiveness and efficiency of clinical trials? clinical trials.2 The current process for developing Adaptive clinical trials enable new treatments is expensive, takes researchers to change an aspect a long time and in some cases, the of a trial design at an interim development process has to be assessment, while controlling stopped after significant amounts the rate of type 1 errors.1 Interim of time and resources have been assessments can help to determine invested.2 In 2006, the FDA published whether a trial design is the most a “Critical Path Opportunities -

Quasi-Experimental Studies in the Fields of Infection Control and Antibiotic Resistance, Ten Years Later: a Systematic Review
HHS Public Access Author manuscript Author ManuscriptAuthor Manuscript Author Infect Control Manuscript Author Hosp Epidemiol Manuscript Author . Author manuscript; available in PMC 2019 November 12. Published in final edited form as: Infect Control Hosp Epidemiol. 2018 February ; 39(2): 170–176. doi:10.1017/ice.2017.296. Quasi-experimental Studies in the Fields of Infection Control and Antibiotic Resistance, Ten Years Later: A Systematic Review Rotana Alsaggaf, MS, Lyndsay M. O’Hara, PhD, MPH, Kristen A. Stafford, PhD, MPH, Surbhi Leekha, MBBS, MPH, Anthony D. Harris, MD, MPH, CDC Prevention Epicenters Program Department of Epidemiology and Public Health, University of Maryland School of Medicine, Baltimore, Maryland. Abstract OBJECTIVE.—A systematic review of quasi-experimental studies in the field of infectious diseases was published in 2005. The aim of this study was to assess improvements in the design and reporting of quasi-experiments 10 years after the initial review. We also aimed to report the statistical methods used to analyze quasi-experimental data. DESIGN.—Systematic review of articles published from January 1, 2013, to December 31, 2014, in 4 major infectious disease journals. METHODS.—Quasi-experimental studies focused on infection control and antibiotic resistance were identified and classified based on 4 criteria: (1) type of quasi-experimental design used, (2) justification of the use of the design, (3) use of correct nomenclature to describe the design, and (4) statistical methods used. RESULTS.—Of 2,600 articles, 173 (7%) featured a quasi-experimental design, compared to 73 of 2,320 articles (3%) in the previous review (P<.01). Moreover, 21 articles (12%) utilized a study design with a control group; 6 (3.5%) justified the use of a quasi-experimental design; and 68 (39%) identified their design using the correct nomenclature. -

Impact of Blinding on Estimated Treatment Effects in Randomised Clinical Trials
RESEARCH Impact of blinding on estimated treatment effects in randomised BMJ: first published as 10.1136/bmj.l6802 on 21 January 2020. Downloaded from clinical trials: meta-epidemiological study Helene Moustgaard,1-4 Gemma L Clayton,5 Hayley E Jones,5 Isabelle Boutron,6 Lars Jørgensen,4 David R T Laursen,1-4 Mette F Olsen,4 Asger Paludan-Müller,4 Philippe Ravaud,6 5,7 5,7,8 5,7 1-3 Jelena Savović, , Jonathan A C Sterne, Julian P T Higgins, Asbjørn Hróbjartsson For numbered affiliations see ABSTRACT 1 indicated exaggerated effect estimates in trials end of the article. OBJECTIVES without blinding. Correspondence to: To study the impact of blinding on estimated RESULTS H Moustgaard treatment effects, and their variation between [email protected] The study included 142 meta-analyses (1153 trials). (or @HeleneMoustgaa1 on Twitter trials; differentiating between blinding of patients, The ROR for lack of blinding of patients was 0.91 ORCID 0000-0002-7057-5251) healthcare providers, and observers; detection bias (95% credible interval 0.61 to 1.34) in 18 meta- Additional material is published and performance bias; and types of outcome (the analyses with patient reported outcomes, and 0.98 online only. To view please visit MetaBLIND study). the journal online. (0.69 to 1.39) in 14 meta-analyses with outcomes C ite this as: BMJ 2020;368:l6802 DESIGN reported by blinded observers. The ROR for lack of http://dx.doi.org/10.1136/bmj.l6802 Meta-epidemiological study. blinding of healthcare providers was 1.01 (0.84 to Accepted: 19 November 2019 DATA SOURCE 1.19) in 29 meta-analyses with healthcare provider Cochrane Database of Systematic Reviews (2013-14). -

Internal and External Validity: Can You Apply Research Study Results to Your Patients? Cecilia Maria Patino1,2,A, Juliana Carvalho Ferreira1,3,B
J Bras Pneumol. 2018;44(3):183-183 CONTINUING EDUCATION: http://dx.doi.org/10.1590/S1806-37562018000000164 SCIENTIFIC METHODOLOGY Internal and external validity: can you apply research study results to your patients? Cecilia Maria Patino1,2,a, Juliana Carvalho Ferreira1,3,b CLINICAL SCENARIO extent to which the results of a study are generalizable to patients in our daily practice, especially for the population In a multicenter study in France, investigators conducted that the sample is thought to represent. a randomized controlled trial to test the effect of prone vs. supine positioning ventilation on mortality among patients Lack of internal validity implies that the results of the with early, severe ARDS. They showed that prolonged study deviate from the truth, and, therefore, we cannot prone-positioning ventilation decreased 28-day mortality draw any conclusions; hence, if the results of a trial are [hazard ratio (HR) = 0.39; 95% CI: 0.25-0.63].(1) not internally valid, external validity is irrelevant.(2) Lack of external validity implies that the results of the trial may not apply to patients who differ from the study STUDY VALIDITY population and, consequently, could lead to low adoption The validity of a research study refers to how well the of the treatment tested in the trial by other clinicians. results among the study participants represent true fi ndings among similar individuals outside the study. INCREASING VALIDITY OF RESEARCH This concept of validity applies to all types of clinical STUDIES studies, including those about prevalence, associations, interventions, and diagnosis. The validity of a research To increase internal validity, investigators should ensure study includes two domains: internal and external validity. -

Development of Validated Instruments
Development of Validated Instruments Michelle Tarver, MD, PhD Medical Officer Division of Ophthalmic and Ear, Nose, and Throat Devices Center for Devices and Radiological Health Food and Drug Administration No Financial Conflicts to Disclose 2 Overview • Define Patient Reported Outcomes (PROs) • Factors to Consider when Developing PROs • FDA Guidance for PROs • Use of PROs in FDA Clinical Trials 3 Patient Reported Outcomes (PROs) • Any report of the status of a patient’s health condition that comes directly from the patient, without interpretation of the patient’s response by a clinician or anyone else • Can be measured in absolute terms (e.g., severity of a symptom) or as a change from a previous measure • In trials, measures the effect of a medical intervention on one or more concepts – Concept is the thing being measured (e.g., symptom, effects on function, severity of health condition) 4 Concepts a PRO May Capture • Symptoms • Symptom impact and functioning • Disability/handicap • Adverse events • Treatment tolerability • Treatment satisfaction • Health-related quality of life 5 Criteria to Consider in PRO Development • Appropriateness – Does the content address the relevant questions for the device? • Acceptability – Is the questionnaire acceptable to patients? • Feasibility – Is it easy to administer and process/analyze? • Interpretability – Are the scores interpretable? Abstracted from (1) Patient Reported Outcomes Measurement Group: University of Oxford (2) NIH PROMIS Instrument Development and Validation Standards 6 Criteria -

Title: a TRANSMISSION-VIRULENCE EVOLUTIONARY TRADE-OFF
1 Title: A TRANSMISSION-VIRULENCE EVOLUTIONARY TRADE-OFF EXPLAINS 2 ATTENUATION OF HIV-1 IN UGANDA 3 Short title: EVOLUTION OF VIRULENCE IN HIV 4 François Blanquart1, Mary Kate Grabowski2, Joshua Herbeck3, Fred Nalugoda4, David 5 Serwadda4,5, Michael A. Eller6, 7, Merlin L. Robb6,7, Ronald Gray2,4, Godfrey Kigozi4, Oliver 6 Laeyendecker8, Katrina A. Lythgoe1,9, Gertrude Nakigozi4, Thomas C. Quinn8, Steven J. 7 Reynolds8, Maria J. Wawer2, Christophe Fraser1 8 1. MRC Centre for Outbreak Analysis and Modelling, Department of Infectious Disease 9 Epidemiology, School of Public Health, Imperial College London, United Kingdom 10 2. Department of Epidemiology, Bloomberg School of Public Health, Johns Hopkins University, 11 Baltimore, MD, USA 12 3. International Clinical Research Center, Department of Global Health, University of 13 Washington, Seattle, WA, USA 14 4. Rakai Health Sciences Program, Entebbe, Uganda 15 5. School of Public Health, Makerere University, Kampala, Uganda 16 6. U.S. Military HIV Research Program, Walter Reed Army Institute of Research, Silver Spring, 17 MD, USA 18 7. Henry M. Jackson Foundation for the Advancement of Military Medicine, Bethesda, MD, USA 19 8. Laboratory of Immunoregulation, Division of Intramural Research, National Institute of 20 Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD, USA 21 9. Department of Zoology, University of Oxford, United Kingdom 22 23 Abstract 24 Evolutionary theory hypothesizes that intermediate virulence maximizes pathogen fitness as 25 a result of a trade-off between virulence and transmission, but empirical evidence remains scarce. 26 We bridge this gap using data from a large and long-standing HIV-1 prospective cohort, in 27 Uganda. -

Development of a Person-Centered Conceptual Model of Perceived Fatigability
Quality of Life Research (2019) 28:1337–1347 https://doi.org/10.1007/s11136-018-2093-z Development of a person-centered conceptual model of perceived fatigability Anna L. Kratz1 · Susan L. Murphy2 · Tiffany J. Braley3 · Neil Basu4 · Shubhangi Kulkarni1 · Jenna Russell1 · Noelle E. Carlozzi1 Accepted: 17 December 2018 / Published online: 2 January 2019 © Springer Nature Switzerland AG 2019 Abstract Purpose Perceived fatigability, reflective of changes in fatigue intensity in the context of activity, has emerged as a poten- tially important clinical outcome and quality of life indicator. Unfortunately, the nature of perceived fatigability is not well characterized. The aim of this study is to define the characteristics of fatigability through the development of a conceptual model informed by input from key stakeholders who experience fatigability, including the general population, individuals with multiple sclerosis (MS), and individuals with fibromyalgia (FM). Methods Thirteen focus groups were conducted with 101 participants; five groups with n = 44 individuals representing the general population, four groups with n = 26 individuals with MS, and four groups with n = 31 individuals with FM. Focus group data were qualitatively analyzed to identify major themes in the participants’ characterizations of perceived fatigability. Results Seven major themes were identified: general fatigability, physical fatigability, mental fatigability, emotional fati- gability, moderators of fatigability, proactive and reactive behaviors, and temporal aspects of fatigability. Relative to those in the general sample, FM or MS groups more often described experiencing fatigue as a result of cognitive activity, use of proactive behaviors to manage fatigability, and sensory stimulation as exacerbating fatigability. Conclusions Fatigability is the complex and dynamic process of the development of physical, mental, and/or emotional fatigue. -

Catalogue of Clinical Trials and Cohort Studies to Identify Biological
Catalogue of clinical trials and cohort studies to identify biological specimens of relevance to the development of assays for acute and early HIV infection: Final Report Catalogue of clinical trials and cohort studies to identify biological specimens of relevance to the development of assays for recent HIV infection Final Report February 2010 This final report was prepared by Dr Joanne Micallef and Professor John Kaldor, The University of New South Wales, under subcontract with Family Health International, funded by the Bill and Melinda Gates Foundation under the grant titled “Development of Assays for Acute HIV Infection and Estimation and HIV Incidence in Population”. 1 Catalogue of clinical trials and cohort studies to identify biological specimens of relevance to the development of assays for acute and early HIV infection: Final Report Table of Contents Table of Contents .................................................................................................................................................................... 2 List of acronyms ...................................................................................................................................................................... 4 1 Introduction ..................................................................................................................................................................... 6 2 Methods ............................................................................................................................................................................ -

Observational Clinical Research
E REVIEW ARTICLE Clinical Research Methodology 2: Observational Clinical Research Daniel I. Sessler, MD, and Peter B. Imrey, PhD * † Case-control and cohort studies are invaluable research tools and provide the strongest fea- sible research designs for addressing some questions. Case-control studies usually involve retrospective data collection. Cohort studies can involve retrospective, ambidirectional, or prospective data collection. Observational studies are subject to errors attributable to selec- tion bias, confounding, measurement bias, and reverse causation—in addition to errors of chance. Confounding can be statistically controlled to the extent that potential factors are known and accurately measured, but, in practice, bias and unknown confounders usually remain additional potential sources of error, often of unknown magnitude and clinical impact. Causality—the most clinically useful relation between exposure and outcome—can rarely be defnitively determined from observational studies because intentional, controlled manipu- lations of exposures are not involved. In this article, we review several types of observa- tional clinical research: case series, comparative case-control and cohort studies, and hybrid designs in which case-control analyses are performed on selected members of cohorts. We also discuss the analytic issues that arise when groups to be compared in an observational study, such as patients receiving different therapies, are not comparable in other respects. (Anesth Analg 2015;121:1043–51) bservational clinical studies are attractive because Group, and the American Society of Anesthesiologists they are relatively inexpensive and, perhaps more Anesthesia Quality Institute. importantly, can be performed quickly if the required Recent retrospective perioperative studies include data O 1,2 data are already available. -

(EPOC) Data Collection Checklist
Cochrane Effective Practice and Organisation of Care Review Group DATA COLLECTION CHECKLIST Page 2 Cochrane Effective Practice and Organisation of Care Review Group (EPOC) Data Collection Checklist CONTENTS Item Page Introduction 5-6 1 Inclusion criteria* 7-8 1.1 Study design* 7 1.1.1 Randomised controlled trial* 1.1.2 Controlled clinical trial* 1.1.3 Controlled before and after study* 1.1.4 Interrupted time series* 1.2 Methodological inclusion criteria* 8 2 Interventions* 9-12 2.1 Type of intervention 9 2.1.1 Professional interventions* 9 2.1.2 Financial interventions* 10 2.1.2.1 Provider interventions* 2.1.2.2 Patient interventions* 2.1.3 Organisational interventions* 11 2.1.3.1 Provider orientated interventions* 2.1.3.2 Patient orientated interventions* 2.1.3.3 Structural interventions* 2.1.4 Regulatory interventions* 12 2.2 Controls* 13 3 Type of targeted behaviour* 13 4 Participants* 14-15 4.1 Characteristics of participating providers* 14 4.1.1 Profession* Item Page Page 3 4.1.2 Level of training* 4.1.3 Clinical speciality* 4.1.4 Age 4.1.5 Time since graduation 4.2 Characteristics of participating patients* 15 4.2.1 Clinical problem* 4.2.2 Other patient characteristics 4.2.3 Number of patients included in the study* 5 Setting* 16 5.1 Reimbursement system 5.2 Location of care* 5.3 Academic Status* 5.4 Country* 5.5 Proportion of eligible providers from the sampling frame* 6 Methods* 17 6.1 Unit of allocation* 6.2 Unit of analysis* 6.3 Power calculation* 6.4 Quality criteria* 17-22 6.4.1 Quality criteria for randomised controlled trials -

Adaptive Designs in Clinical Trials: Why Use Them, and How to Run and Report Them Philip Pallmann1* , Alun W
Pallmann et al. BMC Medicine (2018) 16:29 https://doi.org/10.1186/s12916-018-1017-7 CORRESPONDENCE Open Access Adaptive designs in clinical trials: why use them, and how to run and report them Philip Pallmann1* , Alun W. Bedding2, Babak Choodari-Oskooei3, Munyaradzi Dimairo4,LauraFlight5, Lisa V. Hampson1,6, Jane Holmes7, Adrian P. Mander8, Lang’o Odondi7, Matthew R. Sydes3,SofíaS.Villar8, James M. S. Wason8,9, Christopher J. Weir10, Graham M. Wheeler8,11, Christina Yap12 and Thomas Jaki1 Abstract Adaptive designs can make clinical trials more flexible by utilising results accumulating in the trial to modify the trial’s course in accordance with pre-specified rules. Trials with an adaptive design are often more efficient, informative and ethical than trials with a traditional fixed design since they often make better use of resources such as time and money, and might require fewer participants. Adaptive designs can be applied across all phases of clinical research, from early-phase dose escalation to confirmatory trials. The pace of the uptake of adaptive designs in clinical research, however, has remained well behind that of the statistical literature introducing new methods and highlighting their potential advantages. We speculate that one factor contributing to this is that the full range of adaptations available to trial designs, as well as their goals, advantages and limitations, remains unfamiliar to many parts of the clinical community. Additionally, the term adaptive design has been misleadingly used as an all-encompassing label to refer to certain methods that could be deemed controversial or that have been inadequately implemented. We believe that even if the planning and analysis of a trial is undertaken by an expert statistician, it is essential that the investigators understand the implications of using an adaptive design, for example, what the practical challenges are, what can (and cannot) be inferred from the results of such a trial, and how to report and communicate the results.