Tutorial Section Entrez: Making Use of Its Power
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Expressed Sequence Tag Analysis of the Response of Apple
Physiologia Plantarum 133: 298–317. 2008 Copyright ª Physiologia Plantarum 2008, ISSN 0031-9317 Expressed sequence tag analysis of the response of apple (Malus x domestica ‘Royal Gala’) to low temperature and water deficit Michael Wisniewskia,*, Carole Bassetta,*, John Norellia, Dumitru Macarisina, Timothy Artlipa, Ksenija Gasicb and Schuyler Korbanb aUnited States Department of Agriculture – Agricultural Research Service (USDA-ARS), Appalachian Fruit Research Station, 2217 Wiltshire Road, Kearneysville, WV 25430, USA bDepartment of Natural Resources and Environmental Sciences, University of Illinois, 310 ERML, 1201 W. Gregory Drive, Urbana, IL 61801, USA Correspondence Leaf, bark, xylem and root tissues were used to make nine cDNA libraries from *Corresponding author, non-stressed (control) ‘Royal Gala’ apple trees, and from ‘Royal Gala’ trees e-mail: [email protected]; exposed to either low temperature (5°C for 24 h) or water deficit (45% of [email protected] saturated pot mass for 2 weeks). Over 22 600 clones from the nine libraries # Received 26 September 2007; revised 3 were subjected to 5 single-pass sequencing, clustered and annotated using January 2008 BLASTX. The number of clusters in the libraries ranged from 170 to 1430. Regarding annotation of the sequences, BLASTX analysis indicated that within doi: 10.1111/j.1399-3054.2008.01063.x the libraries 65–72% of the clones had a high similarity to known function genes, 6–15% had no functional assignment and 15–26% were completely novel. The expressed sequence tags were combined into three classes (control, low-temperature and water deficit) and the annotated genes in each class were placed into 1 of 10 different functional categories. -

HEREDITARY CANCER PANELS Part I
Pathology and Laboratory Medicine Clinic Building, K6, Core Lab, E-655 2799 W. Grand Blvd. HEREDITARY CANCER PANELS Detroit, MI 48202 855.916.4DNA (4362) Part I- REQUISITION Required Patient Information Ordering Physician Information Name: _________________________________________________ Gender: M F Name: _____________________________________________________________ MRN: _________________________ DOB: _______MM / _______DD / _______YYYY Address: ___________________________________________________________ ICD10 Code(s): _________________/_________________/_________________ City: _______________________________ State: ________ Zip: __________ ICD-10 Codes are required for billing. When ordering tests for which reimbursement will be sought, order only those tests that are medically necessary for the diagnosis and treatment of the patient. Phone: _________________________ Fax: ___________________________ Billing & Collection Information NPI: _____________________________________ Patient Demographic/Billing/Insurance Form is required to be submitted with this form. Most genetic testing requires insurance prior authorization. Due to high insurance deductibles and member policy benefits, patients may elect to self-pay. Call for more information (855.916.4362) Bill Client or Institution Client Name: ______________________________________________________ Client Code/Number: _____________ Bill Insurance Prior authorization or reference number: __________________________________________ Patient Self-Pay Call for pricing and payment options Toll -

Impact of the Protein Data Bank Across Scientific Disciplines.Data Science Journal, 19: 25, Pp
Feng, Z, et al. 2020. Impact of the Protein Data Bank Across Scientific Disciplines. Data Science Journal, 19: 25, pp. 1–14. DOI: https://doi.org/10.5334/dsj-2020-025 RESEARCH PAPER Impact of the Protein Data Bank Across Scientific Disciplines Zukang Feng1,2, Natalie Verdiguel3, Luigi Di Costanzo1,4, David S. Goodsell1,5, John D. Westbrook1,2, Stephen K. Burley1,2,6,7,8 and Christine Zardecki1,2 1 Research Collaboratory for Structural Bioinformatics Protein Data Bank, Rutgers, The State University of New Jersey, Piscataway, NJ, US 2 Institute for Quantitative Biomedicine, Rutgers, The State University of New Jersey, Piscataway, NJ, US 3 University of Central Florida, Orlando, Florida, US 4 Department of Agricultural Sciences, University of Naples Federico II, Portici, IT 5 Department of Integrative Structural and Computational Biology, The Scripps Research Institute, La Jolla, CA, US 6 Research Collaboratory for Structural Bioinformatics Protein Data Bank, San Diego Supercomputer Center, University of California, San Diego, La Jolla, CA, US 7 Rutgers Cancer Institute of New Jersey, Rutgers, The State University of New Jersey, New Brunswick, NJ, US 8 Skaggs School of Pharmacy and Pharmaceutical Sciences, University of California, San Diego, La Jolla, CA, US Corresponding author: Christine Zardecki ([email protected]) The Protein Data Bank archive (PDB) was established in 1971 as the 1st open access digital data resource for biology and medicine. Today, the PDB contains >160,000 atomic-level, experimentally-determined 3D biomolecular structures. PDB data are freely and publicly available for download, without restrictions. Each entry contains summary information about the structure and experiment, atomic coordinates, and in most cases, a citation to a corresponding scien- tific publication. -

Mismatch Repair Gene PMS2: Disease-Causing Germline
[CANCER RESEARCH 64, 4721–4727, July 15, 2004] Mismatch Repair Gene PMS2: Disease-Causing Germline Mutations Are Frequent in Patients Whose Tumors Stain Negative for PMS2 Protein, but Paralogous Genes Obscure Mutation Detection and Interpretation Hidewaki Nakagawa,1 Janet C. Lockman,1 Wendy L. Frankel,2 Heather Hampel,1 Kelle Steenblock,3 Lawrence J. Burgart,3 Stephen N. Thibodeau,3 and Albert de la Chapelle1 1Division of Human Cancer Genetics, Comprehensive Cancer Center, and 2Department of Surgical Pathology, The Ohio State University, Columbus, Ohio; and 3Departments of Laboratory Medicine and Pathology, Mayo Clinic and Foundation, Rochester, Minnesota ABSTRACT derived from a family described by Trimbath et al. (3). In two additional cases, children with cancer were heterozygous for germline ␣ The MutL heterodimer formed by mismatch repair (MMR) proteins mutations and even showed widespread microsatellite instability in MLH1 and PMS2 is a major component of the MMR complex, yet normal tissues, but the evidence appeared to support the notion of mutations in the PMS2 gene are rare in the etiology of hereditary non- polyposis colorectal cancer. Evidence from five published cases suggested recessive inheritance (even though a second mutation was not found), that contrary to the Knudson principle, PMS2 mutations cause hereditary in that a parent who had the same mutation had no cancer (4, 5). The nonpolyposis colorectal cancer or Turcot syndrome only when they are fifth patient reported was heterozygous for a PMS2 germline muta- biallelic in the germline or abnormally expressed. As candidates for PMS2 tion, but clinical features were not described (6). mutations, we selected seven patients whose colon tumors stained negative The Knudson two-hit model (7) applies to the MLH1, MSH2, and for PMS2 and positive for MLH1 by immunohistochemistry. -

Deficiency in DNA Mismatch Repair of Methylation Damage Is a Major
bioRxiv preprint doi: https://doi.org/10.1101/2020.11.18.388108; this version posted November 18, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Deficiency in DNA mismatch repair of methylation damage is a major 2 mutational process in cancer 3 1 1 1 1 1,* 4 Hu Fang , Xiaoqiang Zhu , Jieun Oh , Jayne A. Barbour , Jason W. H. Wong 5 6 1School of Biomedical Sciences, Li Ka Shing Faculty of Medicine, The University of 7 Hong Kong, Hong Kong Special Administrative Region 8 9 *Correspondence: [email protected] 10 11 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.11.18.388108; this version posted November 18, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Abstract 2 DNA mismatch repair (MMR) is essential for maintaining genome integrity with its 3 deficiency predisposing to cancer1. MMR is well known for its role in the post- 4 replicative repair of mismatched base pairs that escape proofreading by DNA 5 polymerases following cell division2. Yet, cancer genome sequencing has revealed that 6 MMR deficient cancers not only have high mutation burden but also harbour multiple 7 mutational signatures3, suggesting that MMR has pleotropic effects on DNA repair. The 8 mechanisms underlying these mutational signatures have remained unclear despite 9 studies using a range of in vitro4,5 and in vivo6 models of MMR deficiency. -

Involvement of MBD4 Inactivation in Mismatch Repair-Deficient Tumorigenesis
Involvement of MBD4 inactivation in mismatch repair-deficient tumorigenesis The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Tricarico, R., S. Cortellino, A. Riccio, S. Jagmohan-Changur, H. van der Klift, J. Wijnen, D. Turner, et al. 2015. “Involvement of MBD4 inactivation in mismatch repair-deficient tumorigenesis.” Oncotarget 6 (40): 42892-42904. Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:26318553 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA www.impactjournals.com/oncotarget/ Oncotarget, Vol. 6, No. 40 Involvement of MBD4 inactivation in mismatch repair-deficient tumorigenesis Rossella Tricarico1, Salvatore Cortellino2, Antonio Riccio3, Shantie Jagmohan- Changur4, Heleen Van der Klift5, Juul Wijnen5, David Turner6, Andrea Ventura7, Valentina Rovella8, Antonio Percesepe9, Emanuela Lucci-Cordisco10, Paolo Radice11, Lucio Bertario11, Monica Pedroni12, Maurizio Ponz de Leon12, Pietro Mancuso1,13, Karthik Devarajan14, Kathy Q. Cai15, Andres J.P. Klein-Szanto15, Giovanni Neri10, Pål Møller16, Alessandra Viel17, Maurizio Genuardi10, Riccardo Fodde4, Alfonso Bellacosa1 1Cancer Epigenetics and Cancer Biology Programs, Fox Chase Cancer Center, Philadelphia, Pennsylvania, United States 2IFOM-FIRC Institute of Molecular Oncology, Milan, -

The Genetics of Bipolar Disorder
Molecular Psychiatry (2008) 13, 742–771 & 2008 Nature Publishing Group All rights reserved 1359-4184/08 $30.00 www.nature.com/mp FEATURE REVIEW The genetics of bipolar disorder: genome ‘hot regions,’ genes, new potential candidates and future directions A Serretti and L Mandelli Institute of Psychiatry, University of Bologna, Bologna, Italy Bipolar disorder (BP) is a complex disorder caused by a number of liability genes interacting with the environment. In recent years, a large number of linkage and association studies have been conducted producing an extremely large number of findings often not replicated or partially replicated. Further, results from linkage and association studies are not always easily comparable. Unfortunately, at present a comprehensive coverage of available evidence is still lacking. In the present paper, we summarized results obtained from both linkage and association studies in BP. Further, we indicated new potential interesting genes, located in genome ‘hot regions’ for BP and being expressed in the brain. We reviewed published studies on the subject till December 2007. We precisely localized regions where positive linkage has been found, by the NCBI Map viewer (http://www.ncbi.nlm.nih.gov/mapview/); further, we identified genes located in interesting areas and expressed in the brain, by the Entrez gene, Unigene databases (http://www.ncbi.nlm.nih.gov/entrez/) and Human Protein Reference Database (http://www.hprd.org); these genes could be of interest in future investigations. The review of association studies gave interesting results, as a number of genes seem to be definitively involved in BP, such as SLC6A4, TPH2, DRD4, SLC6A3, DAOA, DTNBP1, NRG1, DISC1 and BDNF. -

Predisposition to Hematologic Malignancies in Patients With
LETTERS TO THE EDITOR carcinomas but no internal cancer by the age of 29 years Predisposition to hematologic malignancies in and 9 years, respectively. patients with xeroderma pigmentosum Case XP540BE . This patient had a highly unusual pres - entation of MPAL. She was diagnosed with XP at the age Germline predisposition is a contributing etiology of of 18 months with numerous lentigines on sun-exposed hematologic malignancies, especially in children and skin, when her family emigrated from Morocco to the young adults. Germline predisposition in myeloid neo - USA. The homozygous North African XPC founder muta - plasms was added to the World Health Organization tion was present. 10 She had her first skin cancer at the age 1 2016 classification, and current management recommen - of 8 years, and subsequently developed more than 40 cuta - dations emphasize the importance of screening appropri - neous basal and squamous cell carcinomas, one melanoma 2 ate patients. Rare syndromes of DNA repair defects can in situ , and one ocular surface squamous neoplasm. She 3 lead to myeloid and/or lymphoid neoplasms. Here, we was diagnosed with a multinodular goiter at the age of 9 describe our experience with hematologic neoplasms in years eight months, with several complex nodules leading the defective DNA repair syndrome, xeroderma pigmen - to removal of her thyroid gland. Histopathology showed tosum (XP), including myelodysplastic syndrome (MDS), multinodular adenomatous/papillary hyperplasia. At the secondary acute myeloid leukemia (AML), high-grade age of 19 years, she presented with night sweats, fatigue, lymphoma, and an extremely unusual presentation of and lymphadenopathy. Laboratory studies revealed pancy - mixed phenotype acute leukemia (MPAL) with B, T and topenia with hemoglobin 6.8 g/dL, platelet count myeloid blasts. -
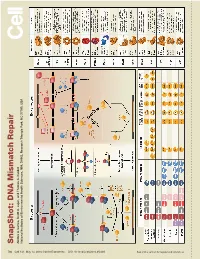
S Na P S H O T: D N a Mism a Tc H R E P a Ir
SnapShot: Repair DNA Mismatch Scott A. Lujan, and Thomas Kunkel A. Larrea, Andres Park, NC 27709, USA Triangle Health Sciences, NIH, DHHS, Research National Institutes of Environmental 730 Cell 141, May 14, 2010 ©2010 Elsevier Inc. DOI 10.1016/j.cell.2010.05.002 See online version for legend and references. SnapShot: DNA Mismatch Repair Andres A. Larrea, Scott A. Lujan, and Thomas A. Kunkel National Institutes of Environmental Health Sciences, NIH, DHHS, Research Triangle Park, NC 27709, USA Mismatch Repair in Bacteria and Eukaryotes Mismatch repair in the bacterium Escherichia coli is initiated when a homodimer of MutS binds as an asymmetric clamp to DNA containing a variety of base-base and insertion-deletion mismatches. The MutL homodimer then couples MutS recognition to the signal that distinguishes between the template and nascent DNA strands. In E. coli, the lack of adenine methylation, catalyzed by the DNA adenine methyltransferase (Dam) in newly synthesized GATC sequences, allows E. coli MutH to cleave the nascent strand. The resulting nick is used for mismatch removal involving the UvrD helicase, single-strand DNA-binding protein (SSB), and excision by single-stranded DNA exonucleases from either direction, depending upon the polarity of the nick relative to the mismatch. DNA polymerase III correctly resynthesizes DNA and ligase completes repair. In bacteria lacking Dam/MutH, as in eukaryotes, the signal for strand discrimination is uncertain but may be the DNA ends associated with replication forks. In these bacteria, MutL harbors a nick-dependent endonuclease that creates a nick that can be used for mismatch excision. Eukaryotic mismatch repair is similar, although it involves several dif- ferent MutS and MutL homologs: MutSα (MSH2/MSH6) recognizes single base-base mismatches and 1–2 base insertion/deletions; MutSβ (MSH2/MSH3) recognizes insertion/ deletion mismatches containing two or more extra bases. -

Pdbefold Tutorial Tutorial Pdbefold Can May Be Accessed from Multiple Locations on the Pdbe Website
PDBe TUTORIAL PDBeFold (SSM: Secondary Structure Matching) http://pdbe.org/fold/ This PDBe tutorial introduces PDBeFold, an interactive service for comparing protein structures in 3D. This service provides: . Pairwise and multiple comparison and 3D alignment of protein structures . Examination of a protein structure for similarity with the whole Protein Data Bank (PDB) archive or SCOP. Best C -alignment of compared structures . Download and visualisation of best-superposed structures using various graphical packages PDBeFold structure alignment is based on identification of residues occupying “equivalent” geometrical positions. In other words, unlike sequence alignment, residue type is neglected. The PDBeFold service is a very powerful structure alignment tool which can perform both pairwise and multiple three dimensional alignment. In addition to this there are various options by which the results of the structural alignment query can be sorted. The results of the Secondary Structure Matching can be sorted based on the Q score (Cα- alignment), P score (taking into account RMSD, number of aligned residues, number of gaps, number of matched Secondary Structure Elements and the SSE match score), Z score (based on Gaussian Statistics), RMSD and % Sequence Identity. It is hoped that at the end of this tutorial users will be able to use PDbeFold for the analysis of their own uploaded structures or entries already in the PDB archive. Protein Data Bank in Europe http://pdbe.org PDBeFOLD Tutorial Tutorial PDBeFold can may be accessed from multiple locations on the PDBe website. From the PDBe home page (http://pdbe.org/), there are two access points for the program as shown below. -

DNA Mismatch Repair Proteins MLH1 and PMS2 Can Be Imported to the Nucleus by a Classical Nuclear Import Pathway
Accepted Manuscript DNA mismatch repair proteins MLH1 and PMS2 can be imported to the nucleus by a classical nuclear import pathway Andrea C. de Barros, Agnes A.S. Takeda, Thiago R. Dreyer, Adrian Velazquez- Campoy, Boštjan Kobe, Marcos R.M. Fontes PII: S0300-9084(17)30308-5 DOI: 10.1016/j.biochi.2017.11.013 Reference: BIOCHI 5321 To appear in: Biochimie Received Date: 25 May 2017 Accepted Date: 22 November 2017 Please cite this article as: Keplinger M, Marhofer P, Moriggl B, Zeitlinger M, Muehleder-Matterey S, Marhofer D, Cutaneous innervation of the hand: clinical testing in volunteers shows that variability is far greater than claimed in textbooks, British Journal of Anaesthesia (2017), doi: 10.1016/j.bja.2017.09.008. This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain. ACCEPTED MANUSCRIPT ABSTRACT MLH1 and PMS2 proteins form the MutL α heterodimer, which plays a major role in DNA mismatch repair (MMR) in humans. Mutations in MMR-related proteins are associated with cancer, especially with colon cancer. The N-terminal region of MutL α comprises the N-termini of PMS2 and MLH1 and, similarly, the C-terminal region of MutL α is composed by the C-termini of PMS2 and MLH1, and the two are connected by linker region. -

EMBL-EBI-Overview.Pdf
EMBL-EBI Overview EMBL-EBI Overview Welcome Welcome to the European Bioinformatics Institute (EMBL-EBI), a global hub for big data in biology. We promote scientific progress by providing freely available data to the life-science research community, and by conducting exceptional research in computational biology. At EMBL-EBI, we manage public life-science data on a very large scale, offering a rich resource of carefully curated information. We make our data, tools and infrastructure openly available to an increasingly data-driven scientific community, adjusting to the changing needs of our users, researchers, trainees and industry partners. This proactive approach allows us to deliver relevant, up-to-date data and tools to the millions of scientists who depend on our services. We are a founding member of ELIXIR, the European infrastructure for biological information, and are central to global efforts to exchange information, set standards, develop new methods and curate complex information. Our core databases are produced in collaboration with other world leaders including the National Center for Biotechnology Information in the US, the National Institute of Genetics in Japan, SIB Swiss Institute of Bioinformatics and the Wellcome Trust Sanger Institute in the UK. We are also a world leader in computational biology research, and are well integrated with experimental and computational groups on all EMBL sites. Our research programme is highly collaborative and interdisciplinary, regularly producing high-impact works on sequence and structural alignment, genome analysis, basic biological breakthroughs, algorithms and methods of widespread importance. EMBL-EBI is an international treaty organisation, and we serve the global scientific community.