Synthesis of Biologically Active Polycyclic Natural Products and Multifunctional Imaging Probes Tezcan Guney Iowa State University
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

By Exemestane, a Novel Irreversible Aromatase Inhibitor, in Postmenopausal Breast Cancer Patients1
Vol. 4, 2089-2093, September 1998 Clinical Cancer Research 2089 In Vivo Inhibition of Aromatization by Exemestane, a Novel Irreversible Aromatase Inhibitor, in Postmenopausal Breast Cancer Patients1 Jfirgen Geisler, Nick King, Gun Anker, ation aromatase inhibitor AG3 has been used for breast cancer Giorgio Ornati, Enrico Di Salle, treatment for more than two decades (1). Because of substantial side effects associated with AG treatment, several new aro- Per Eystein L#{248}nning,2 and Mitch Dowsett matase inhibitors have been introduced in clinical trials. Department of Oncology, Haukeland University Hospital, N-502l Aromatase inhibitors can be divided into two major classes Bergen, Norway [J. G., G. A., P. E. L.]; Academic Department of Biochemistry, Royal Marsden Hospital, London, SW3 6JJ, United of compounds, steroidal and nonsteroidal drugs. Nonsteroidal Kingdom [N. K., M. D.]; and Department of Experimental aromatase inhibitors include AG and the imidazole/triazole Endocrinology, Pharmacia and Upjohn, 20014 Nerviano, Italy [G. 0., compounds. With the exception of testololactone, a testosterone E. D. S.] derivative (2), steroidal aromatase inhibitors are all derivatives of A, the natural substrate for the aromatase enzyme (3). The second generation steroidal aromatase inhibitor, 4- ABSTRACT hydroxyandrostenedione (4-OHA, formestane), was found to The effect of exemestane (6-methylenandrosta-1,4- inhibit peripheral aromatization by -85% when administered diene-3,17-dione) 25 mg p.o. once daily on in vivo aromati- by the i.m. route at a dosage of 250 mg every 2 weeks as zation was studied in 10 postmenopausal women with ad- recommended (4) but only by 50-70% (5) when administered vanced breast cancer. -

Part I: Carbonyl-Olefin Metathesis of Norbornene
Part I: Carbonyl-Olefin Metathesis of Norbornene Part II: Cyclopropenimine-Catalyzed Asymmetric Michael Reactions Zara Maxine Seibel Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2016 1 © 2016 Zara Maxine Seibel All Rights Reserved 2 ABSTRACT Part I: Carbonyl-Olefin Metathesis of Norbornene Part II: Cyclopropenimine-Catalyzed Asymmetric Michael Reactions Zara Maxine Seibel This thesis details progress towards the development of an organocatalytic carbonyl- olefin metathesis of norbornene. This transformation has not previously been done catalytically and has not been done in practical manner with stepwise or stoichiometric processes. Building on the previous work of the Lambert lab on the metathesis of cyclopropene and an aldehyde using a hydrazine catalyst, this work discusses efforts to expand to the less stained norbornene. Computational and experimental studies on the catalytic cycle are discussed, including detailed experimental work on how various factors affect the difficult cycloreversion step. The second portion of this thesis details the use of chiral cyclopropenimine bases as catalysts for asymmetric Michael reactions. The Lambert lab has previously developed chiral cyclopropenimine bases for glycine imine nucleophiles. The scope of these catalysts was expanded to include glycine imine derivatives in which the nitrogen atom was replaced with a carbon atom, and to include imines derived from other amino acids. i Table of Contents List of Abbreviations…………………………………………………………………………..iv Part I: Carbonyl-Olefin Metathesis…………………………………………………………… 1 Chapter 1 – Metathesis Reactions of Double Bonds………………………………………….. 1 Introduction………………………………………………………………………………. 1 Olefin Metathesis………………………………………………………………………… 2 Wittig Reaction…………………………………………………………………………... 6 Tebbe Olefination………………………………………………………………………... 9 Carbonyl-Olefin Metathesis……………………………………………………………. -

New Cyclophanes As Supramolecular Scaffolds: the Synthesis of Tribenzo-1,4,7-Triazacyclononatriene
Loyola University Chicago Loyola eCommons Dissertations Theses and Dissertations 2010 New Cyclophanes as Supramolecular Scaffolds: The Synthesis of Tribenzo-1,4,7-Triazacyclononatriene. Andria M. Panagopoulos Loyola University Chicago Follow this and additional works at: https://ecommons.luc.edu/luc_diss Part of the Biochemistry Commons Recommended Citation Panagopoulos, Andria M., "New Cyclophanes as Supramolecular Scaffolds: The Synthesis of Tribenzo-1,4,7-Triazacyclononatriene." (2010). Dissertations. 88. https://ecommons.luc.edu/luc_diss/88 This Dissertation is brought to you for free and open access by the Theses and Dissertations at Loyola eCommons. It has been accepted for inclusion in Dissertations by an authorized administrator of Loyola eCommons. For more information, please contact [email protected]. This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 3.0 License. Copyright © 2010 Andria M. Panagopoulos LOYOLA UNIVERSITY CHICAGO NEW CYCLOPHANES AS SUPRAMOLECULAR SCAFFOLDS: THE SYNTHESIS OF TRIBENZO-1,4,7-TRIAZACYCLONONATRIENE A DISSERTATION SUBMITTED TO THE FACULTY OF THE GRADUATE SCHOOL IN CANDIDACY FOR THE DEGREE OF DOCTOR OF PHILOSOPHY PROGRAM IN CHEMISTRY BY ANDRIA M. PANAGOPOULOS CHICAGO, ILLINOIS AUGUST 2010 Copyright by Andria M. Panagopoulos, 2010 All rights reserved ACKNOWLEDGEMENTS I would like to sincerely thank my advisor, Daniel P. Becker, Ph.D. for all his support and guidance throughout the years. I have learned so much from you, not only about chemistry, but about myself and for that I thank you. I would also like to thank my friends and family. You guys have stood by my side, encouraged me and supported me. I could not have completed this without your love and support. -
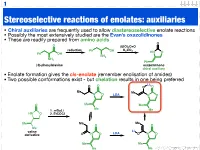
Stereoselective Reactions of Enolates: Auxiliaries
1 Stereoselective reactions of enolates: auxiliaries • Chiral auxiliaries are frequently used to allow diastereoselective enolate reactions • Possibly the most extensively studied are the Evan’s oxazolidinones • These are readily prepared from amino acids O O (EtO)2C=O reduction K CO Ph OH 2 3 HN O Ph OH NH2 NH2 Ph (S)-phenylalanine oxazolidinone chiral auxiliary • Enolate formation gives the cis-enolate (remember enolisation of amides) • Two possible conformations exist - but chelation results in one being preferred Li O O O O Me Me N O LDA N O O Me Me Me 1. n-BuLi Me HN O 2. EtCOCl Me Me Me O O Me Li valine LDA derivative O N O O N O Me Me Me Me 123.702 Organic Chemistry 2 Diastereoselective alkylation of Evan’s enolate Li O O O O Me Me N O PhCH2I N O Ph Me Me Me Me I Ph Li O Bn O O O O O H H Me N Me N H Me Me Me Me iso-propyl group blocks bottom face • Clearly (I hope) one face of the enolate is blocked • Chelation results in a rigid structure that provides maximum steric hindrance • The electrophile can only approach from one face 123.702 Organic Chemistry 3 Diastereoselective alkylation of Evan’s enolate Li O O O O Me Me N O PhCH2I N O Ph Me Me Me Me I Ph Li O Bn O O O O O H H Me N Me N H Me Me Me Me iso-propyl group blocks bottom face • Clearly (I hope) one face of the enolate is blocked • Chelation results in a rigid structure that provides maximum steric hindrance • The electrophile can only approach from one face 123.702 Organic Chemistry 4 Diastereoselective functionalisation Li O O O O Br LDA Me Me N O N O H Ph Ph 96% de H O -

Aromasin (Exemestane)
HIGHLIGHTS OF PRESCRIBING INFORMATION ------------------------------ADVERSE REACTIONS------------------------------ These highlights do not include all the information needed to use • Early breast cancer: Adverse reactions occurring in ≥10% of patients in AROMASIN safely and effectively. See full prescribing information for any treatment group (AROMASIN vs. tamoxifen) were hot flushes AROMASIN. (21.2% vs. 19.9%), fatigue (16.1% vs. 14.7%), arthralgia (14.6% vs. 8.6%), headache (13.1% vs. 10.8%), insomnia (12.4% vs. 8.9%), and AROMASIN® (exemestane) tablets, for oral use increased sweating (11.8% vs. 10.4%). Discontinuation rates due to AEs Initial U.S. Approval: 1999 were similar between AROMASIN and tamoxifen (6.3% vs. 5.1%). Incidences of cardiac ischemic events (myocardial infarction, angina, ----------------------------INDICATIONS AND USAGE--------------------------- and myocardial ischemia) were AROMASIN 1.6%, tamoxifen 0.6%. AROMASIN is an aromatase inhibitor indicated for: Incidence of cardiac failure: AROMASIN 0.4%, tamoxifen 0.3% (6, • adjuvant treatment of postmenopausal women with estrogen-receptor 6.1). positive early breast cancer who have received two to three years of • Advanced breast cancer: Most common adverse reactions were mild to tamoxifen and are switched to AROMASIN for completion of a total of moderate and included hot flushes (13% vs. 5%), nausea (9% vs. 5%), five consecutive years of adjuvant hormonal therapy (14.1). fatigue (8% vs. 10%), increased sweating (4% vs. 8%), and increased • treatment of advanced breast cancer in postmenopausal women whose appetite (3% vs. 6%) for AROMASIN and megestrol acetate, disease has progressed following tamoxifen therapy (14.2). respectively (6, 6.1). ----------------------DOSAGE AND ADMINISTRATION----------------------- To report SUSPECTED ADVERSE REACTIONS, contact Pfizer Inc at Recommended Dose: One 25 mg tablet once daily after a meal (2.1). -

Organic Synthesis: Handout 1
Prof Tim Donohoe: Strategies and Taccs in Organic Synthesis: Handout 1 Organic Synthesis III 8 x 1hr Lectures: Michaelmas Term Weeks 5-8 2016 Mon at 10am; Wed at 9am Dyson Perrins lecture theatre Copies of this handout will be available at hEp://donohoe.chem.ox.ac.uk/page16/index.html 1/33 Prof Tim Donohoe: Strategies and Taccs in Organic Synthesis: Handout 1 Organic Synthesis III Synopsis 1) Introduc5on to synthesis: (i) Why do we want to synthesise molecules- what sort of molecules do we need to make? (ii) What aspects of selecvity do we need to accomplish a good synthesis (chemo-, regio- and stereoselecvity)? (iii) Protecng group chemistry is central to any syntheAc effort (examples and principles) (iv) What is the perfect synthesis (performed in industry versus academia)? 2) The chiral pool: where does absolute stereochemistry come from? 3) Retrosynthesis- learning to think backwards (revision from first and second year). Importance of making C-C bonds and controlling oxidaAon state. Umpolung 4) Some problems to think about 5) Examples of retrosynthesis/synthesis in ac5on. 6) Ten handy hints for retrosynthesis 7) Soluons to the problems Recommended books: General: Organic Chemistry (Warren et al) Organic Synthesis: The DisconnecRon Approach (S. Warren) Classics in Total Synthesis Volumes I and II (K. C. Nicolaou) The Logic of Chemical Synthesis (E. J. Corey) 2/33 View Article Online / Journal Homepage / Table of Contents for this issue 619461 Strychniqae and BYucine. Pavt XLII. 903 Prof Tim Donohoe: Strategies and Taccs in Organic Synthesis: Handout 1 (i) Why do we want to synthesise complex molecules? Isolated from the Pacific Yew in 1962 Prescribed for prostate, breast and ovarian cancer Unique mode of acRon 1x 100 year old tree = 300 mg Taxol Isolated in 1818- poisonous Stuctural elucidaon took R. -

ECO-Ssls for Pahs
Ecological Soil Screening Levels for Polycyclic Aromatic Hydrocarbons (PAHs) Interim Final OSWER Directive 9285.7-78 U.S. Environmental Protection Agency Office of Solid Waste and Emergency Response 1200 Pennsylvania Avenue, N.W. Washington, DC 20460 June 2007 This page intentionally left blank TABLE OF CONTENTS 1.0 INTRODUCTION .......................................................1 2.0 SUMMARY OF ECO-SSLs FOR PAHs......................................1 3.0 ECO-SSL FOR TERRESTRIAL PLANTS....................................4 5.0 ECO-SSL FOR AVIAN WILDLIFE.........................................8 6.0 ECO-SSL FOR MAMMALIAN WILDLIFE..................................8 6.1 Mammalian TRV ...................................................8 6.2 Estimation of Dose and Calculation of the Eco-SSL ........................9 7.0 REFERENCES .........................................................16 7.1 General PAH References ............................................16 7.2 References Used for Derivation of Plant and Soil Invertebrate Eco-SSLs ......17 7.3 References Rejected for Use in Derivation of Plant and Soil Invertebrate Eco-SSLs ...............................................................18 7.4 References Used in Derivation of Wildlife TRVs .........................25 7.5 References Rejected for Use in Derivation of Wildlife TRV ................28 i LIST OF TABLES Table 2.1 PAH Eco-SSLs (mg/kg dry weight in soil) ..............................4 Table 3.1 Plant Toxicity Data - PAHs ..........................................5 Table 4.1 -

IV (Advance Organic Synthesis and Supramolecular Chemistry and Carbocyclic Rings) Module No and 9: Protection and Deprotection Title Module Tag CHE P14 M9
Subject Chemistry Paper No and Title 14: Organic Chemistry –IV (Advance Organic Synthesis and Supramolecular Chemistry and carbocyclic rings) Module No and 9: Protection and deprotection Title Module Tag CHE_P14_M9 CHEMISTRY Paper No. 14: Organic Chemistry –IV (Advance Organic Synthesis and Supramolecular Chemistry and carbocyclic rings) Module No. 9: Protection and deprotection Table of Content 1. Learning outcomes 2. Introduction 3. Protecting groups for carbonyl compounds 4. Protecting groups for alcohols 5. Protecting groups for amines 6. Summary CHEMISTRY Paper No. 14: Organic Chemistry –IV (Advance Organic Synthesis and Supramolecular Chemistry and carbocyclic rings) Module No. 9: Protection and deprotection 1. Learning Outcomes After studying this module, you shall be able to Know about protecting groups. Study the characteristics of protecting groups. Understand the functional group protection. Know the protection of important functional groups. 2. Introduction Protection and deprotection is an important part of organic synthesis. During the course of synthesis, we many times desire to perform reaction at only one of the two functional groups in any single organic molecules. For example, in an organic compound possessing two functional groups like ester and ketone, we have to perform reaction at only ester group, them the keto group needs to be protected. If we want to reduce the ester group, then keto group will also get reduced. To avoid this type of complications, protection and deprotection of functional groups are necessary. 3. Protecting groups for carbonyl compounds The protecting groups allow the masking of a particular functional group where a specified reaction is not to be performed. The protection is required as it interferes with another reaction. -

Enantioselective Total Synthesis of (-)-Deoxoapodine
Enantioselective total synthesis of (-)-deoxoapodine The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Kang, Taek, et al., "Enantioselective total synthesis of (-)- deoxoapodine." Angewandte Chemie International Edition 56, 44 (Sept. 2017): p. 13857-60 doi 10.1002/anie.201708088 ©2017 Author(s) As Published 10.1002/anie.201708088 Publisher Wiley Version Author's final manuscript Citable link https://hdl.handle.net/1721.1/125957 Terms of Use Creative Commons Attribution-Noncommercial-Share Alike Detailed Terms http://creativecommons.org/licenses/by-nc-sa/4.0/ HHS Public Access Author manuscript Author ManuscriptAuthor Manuscript Author Angew Manuscript Author Chem Int Ed Engl Manuscript Author . Author manuscript; available in PMC 2018 October 23. Published in final edited form as: Angew Chem Int Ed Engl. 2017 October 23; 56(44): 13857–13860. doi:10.1002/anie.201708088. Enantioselective Total Synthesis of (−)-Deoxoapodine Dr. Taek Kang§,a, Dr. Kolby L. White§,a, Tyler J. Mannb, Prof. Dr. Amir H. Hoveydab, and Prof. Dr. Mohammad Movassaghia aDepartment of Chemistry, Massachusetts Institute of Technology Cambridge, MA 02139 (USA) bDepartment of Chemistry, Merkert Chemistry Center, Boston College, Chestnut Hill, MA 02467 (USA) Abstract The first enantioselective total synthesis of (−)-deoxoapodine is described. Our synthesis of this hexacyclic aspidosperma alkaloid includes an efficient molybdenum-catalyzed enantioselective ring-closing metathesis reaction for desymmetrization of an advanced intermediate that introduces the C5-quaternary stereocenter. After C21-oxygenation, the pentacyclic core was accessed via an electrophilic C19-amide activation and transannular spirocyclization. A biogenetically inspired dehydrative C6-etherification reaction proved highly effective to secure the F-ring and the fourth contiguous stereocenter of (−)-deoxoapodine with complete stereochemical control. -

Heterocyclic Compounds
Gábor Krajsovszky Heterocyclic compounds ISBN: 978-615-5722-01-1 © Gábor Krajsovszky Responsible editor: Gábor Krajsovszky Publisher’s reader: István Mándity Translated by Péter Tétényi Department of Organic Chemistry Pharmaceutical Faculty Semmelweis University Budapest, 2018 Acknowledgements The editor wants to express many thanks to Dr. István Mándity, who is Associate Professor and Director of Department of Organic Chemistry, for the careful proofreading service of the current manuscript, as well as to Dr. Péter Tétényi, who is Assistant Professor, for the translation to English language. Moreover, the editor renders many thanks to Mrs. Ferenc Juhász and Ms. Nikoletta Zlatzky laboratory assistants for drawing material of the figures. Dr. Gábor Krajsovszky Associate Professor Department of Organic Chemistry Literature used Alan R. Katritzky, Charles W. Rees: Comprehensive Heterocyclic Chemistry Parts 2-3, 4-6, 7 Pergamon Press 1984 Oxford • New York • Toronto • Sydney • Paris • Frankfurt T. Eicher, S. Hauptmann, A. Speicher: The Chemistry of Heterocycles Structure, Reactions, Syntheses, and Applications Wiley-VCH GmbH 2003 Weinheim E. Breitmaier, G. Jung: Organische Chemie Grundlagen, Stoffklassen, Reaktionen, Konzepte, Molekülstruktur Georg Thieme Verlag 1978, 2005 Stuttgart • New York Clauder Ottó: Szerves kémia II/2. Egyetemi jegyzet Semmelweis OTE Budapest, 1980 Bruckner Győző: Szerves kémia III−1. Tankönyvkiadó, Budapest, 1964 Természettudományi Lexikon − Harmadik kötet Clauder Ottó: 'Heterociklusos vegyületek' címszó, 155-161. -

Page 1 of 108 RSC Advances
RSC Advances This is an Accepted Manuscript, which has been through the Royal Society of Chemistry peer review process and has been accepted for publication. Accepted Manuscripts are published online shortly after acceptance, before technical editing, formatting and proof reading. Using this free service, authors can make their results available to the community, in citable form, before we publish the edited article. This Accepted Manuscript will be replaced by the edited, formatted and paginated article as soon as this is available. You can find more information about Accepted Manuscripts in the Information for Authors. Please note that technical editing may introduce minor changes to the text and/or graphics, which may alter content. The journal’s standard Terms & Conditions and the Ethical guidelines still apply. In no event shall the Royal Society of Chemistry be held responsible for any errors or omissions in this Accepted Manuscript or any consequences arising from the use of any information it contains. www.rsc.org/advances Page 1 of 108 RSC Advances Applications of oxazolidinones as chiral auxiliaries in the asymmetric alkylation reaction applied to total synthesis Majid M. Heravi,* Vahideh Zadsirjan, Behnaz Farajpour Department of Chemistry, School of Science, Alzahra University, Vanak, Tehran, Iran Email: [email protected] Abstract Various chiral oxazolidinones (Evans' oxazolidinones) have been employed as effective chiral auxiliaries in the asymmetric alkylation of different enolates. This strategy has been found promising and successful when used as key step (steps) in the total synthesis of several biologically active natural products. In this report, we try to underscore the applications of Manuscript oxazolidinones as chiral auxiliary in asymmetric alkylation, and particularly in crucial chiral inducing steps in the total synthesis of natural products, showing biological activities. -

Poly( Ethylene-Co-Methyl Acrylate)-Solvent- Cosolvent Phase Behaviour at High Pressures
Poly( ethylene-co-methyl acrylate)-solvent- cosolvent phase behaviour at high pressures Melchior A. Meilchen, Bruce M. Hasch, Sang-Ho Lee and Mark A. McHugh* Department of Chemical Engineering, Johns Hopkins University, Baltimore, MD 21278, USA (Received 5 April 7991; accepted 75 July 1997 ) Cloud-point data to 160°C and 2000 bar are presented showing the effect of cosolvents on the phase behaviour of poly(ethylene-co-methyl acrylate) (EMA) (64 mo1%/36 mol%) with propane and chlorodifluoromethane (F22). Ethanol shifts the EMA-propane cloud-point curves to lower temperatures and pressures, but above _ 10 wt% ethanol, the copolymer becomes insoluble. Up to 40 wt% acetone monotonically shifts the EMA-propane cloud-point curves to lower temperatures and pressures. Acetone and ethanol both shift the cloud-point curves of EMA-F22 mixtures in the same monotonic manner for cosolvent concentrations of up to 40 wt%. (Keywords: copolymer; cosolvent; high pressures) INTRODUCTION or diethyl ether, so it is necessary to operate at elevated temperatures and pressures to dissolve high molecular Within the past 20 years there has been a great deal of weight polystyrene in either solvent. Adding acetone to effort invested in trying to understand and model the PS-diethyl ether mixtures monotonically reduces the solubility behaviour of polar polymers in liquid cosolvent pressure needed at a given temperature to obtain a single mixtures’-9. Polymer solubility is usually related to phase. whether the cosolvent preferentially solvates or adsorbs There has also been a number of viscometric and light to certain segments of the polymer chain as determined scattering studies on the solution behaviour of polar by light scattering, viscosity measurements or cloud- copolymers in liquid cosolvent mixtures’4*‘5.