A Multi-Ethnic Meta-Analysis Identifies Novel Genes, Including ACSL5
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

S41467-020-18249-3.Pdf
ARTICLE https://doi.org/10.1038/s41467-020-18249-3 OPEN Pharmacologically reversible zonation-dependent endothelial cell transcriptomic changes with neurodegenerative disease associations in the aged brain Lei Zhao1,2,17, Zhongqi Li 1,2,17, Joaquim S. L. Vong2,3,17, Xinyi Chen1,2, Hei-Ming Lai1,2,4,5,6, Leo Y. C. Yan1,2, Junzhe Huang1,2, Samuel K. H. Sy1,2,7, Xiaoyu Tian 8, Yu Huang 8, Ho Yin Edwin Chan5,9, Hon-Cheong So6,8, ✉ ✉ Wai-Lung Ng 10, Yamei Tang11, Wei-Jye Lin12,13, Vincent C. T. Mok1,5,6,14,15 &HoKo 1,2,4,5,6,8,14,16 1234567890():,; The molecular signatures of cells in the brain have been revealed in unprecedented detail, yet the ageing-associated genome-wide expression changes that may contribute to neurovas- cular dysfunction in neurodegenerative diseases remain elusive. Here, we report zonation- dependent transcriptomic changes in aged mouse brain endothelial cells (ECs), which pro- minently implicate altered immune/cytokine signaling in ECs of all vascular segments, and functional changes impacting the blood–brain barrier (BBB) and glucose/energy metabolism especially in capillary ECs (capECs). An overrepresentation of Alzheimer disease (AD) GWAS genes is evident among the human orthologs of the differentially expressed genes of aged capECs, while comparative analysis revealed a subset of concordantly downregulated, functionally important genes in human AD brains. Treatment with exenatide, a glucagon-like peptide-1 receptor agonist, strongly reverses aged mouse brain EC transcriptomic changes and BBB leakage, with associated attenuation of microglial priming. We thus revealed tran- scriptomic alterations underlying brain EC ageing that are complex yet pharmacologically reversible. -

DYSREGULATION of LONG-CHAIN ACYL-Coa SYNTHETASES in CANCER and THEIR TARGETING STRATEGIES in ANTICANCER THERAPY Md Amir Hossain1, Jun Ma2, Yong Yang1 1
Hossain et al RJLBPCS 2020 www.rjlbpcs.com Life Science Informatics Publications Original Review Article DOI: 10.26479/2020.0603.06 DYSREGULATION OF LONG-CHAIN ACYL-CoA SYNTHETASES IN CANCER AND THEIR TARGETING STRATEGIES IN ANTICANCER THERAPY Md Amir Hossain1, Jun Ma2, Yong Yang1 1. Academic Institute of Pharmaceutical Science, China Pharmaceutical University, Nanjing, China. 2. Ningxia Baoshihua Hospital, Ningxia, China. ABSTRACT: Cancer is a leading cause of death worldwide, and the number of cases globally continues to increase. Cancer is caused by certain changes in genes that are involved in controlling cell functions and cell division. Notably, metabolic dysregulation is one the hallmarks of cancer, and increases in fatty acid metabolism have been demonstrated to promote the growth and survival of a variety of cancers. In human, fatty acids either can be breakdown into acetyl-CoA through catabolic metabolism and aid in ATP generation or in the anabolic metabolism they can incorporate into triacylglycerol and phospholipid. Importantly, both of these pathways need activation of fatty acids, and the key players in this activation of fatty acids are the long-chain acyl-CoA synthetases (ACSLs) that are commonly dysregulated in cancer and associated with oncogenesis and survival. Therefore, it provides a rationale to target ACSLs in cancer. This review summarizes the current understanding of long-chain acyl-CoA synthetases in cancer and their targeting opportunities. KEYWORDS: Acyl-CoA synthetases, cancer, fatty acid, lipid metabolism. Article History: Received: April 15, 2020; Revised: May 10, 2020; Accepted: May 30, 2020. Corresponding Author: Prof. Yong Yang* Academic Institute of Pharmaceutical Science, China Pharmaceutical University, Nanjing, China. -

Aneuploidy: Using Genetic Instability to Preserve a Haploid Genome?
Health Science Campus FINAL APPROVAL OF DISSERTATION Doctor of Philosophy in Biomedical Science (Cancer Biology) Aneuploidy: Using genetic instability to preserve a haploid genome? Submitted by: Ramona Ramdath In partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biomedical Science Examination Committee Signature/Date Major Advisor: David Allison, M.D., Ph.D. Academic James Trempe, Ph.D. Advisory Committee: David Giovanucci, Ph.D. Randall Ruch, Ph.D. Ronald Mellgren, Ph.D. Senior Associate Dean College of Graduate Studies Michael S. Bisesi, Ph.D. Date of Defense: April 10, 2009 Aneuploidy: Using genetic instability to preserve a haploid genome? Ramona Ramdath University of Toledo, Health Science Campus 2009 Dedication I dedicate this dissertation to my grandfather who died of lung cancer two years ago, but who always instilled in us the value and importance of education. And to my mom and sister, both of whom have been pillars of support and stimulating conversations. To my sister, Rehanna, especially- I hope this inspires you to achieve all that you want to in life, academically and otherwise. ii Acknowledgements As we go through these academic journeys, there are so many along the way that make an impact not only on our work, but on our lives as well, and I would like to say a heartfelt thank you to all of those people: My Committee members- Dr. James Trempe, Dr. David Giovanucchi, Dr. Ronald Mellgren and Dr. Randall Ruch for their guidance, suggestions, support and confidence in me. My major advisor- Dr. David Allison, for his constructive criticism and positive reinforcement. -

Flawed Phospholipid Formation Or Faulty Fatty Acid Oxidation: Determining the Cause of Mitochondrial Dysfunction in Hearts Lacking Acsl1
FLAWED PHOSPHOLIPID FORMATION OR FAULTY FATTY ACID OXIDATION: DETERMINING THE CAUSE OF MITOCHONDRIAL DYSFUNCTION IN HEARTS LACKING ACSL1 Trisha J. Grevengoed A dissertation submitted to the faculty at the University of North Carolina at Chapel Hill in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Nutrition (Biochemistry) in the School of Public Health. Chapel Hill 2015 Approved by: Rosalind A. Coleman Stephen D. Hursting Liza Makowski Leslie V. Parise Steven H. Zeisel © 2015 Trisha J. Grevengoed ALL RIGHTS RESERVED ii ABSTRACT Trisha J. Grevengoed: Fatty acid activation in cardiac mitochondria: The role of ACSL1 in phospholipid formation and remodeling, substrate switching, and autophagic flux (Under the direction of Rosalind A. Coleman) Cardiovascular disease is the number one cause of death worldwide. In the heart, mitochondria provide up to 95% of energy, with most of this energy coming from metabolism of fatty acids (FA). FA must be converted to acyl-CoAs by acyl-CoA synthetases (ACS) before entry into pathways of β- oxidation or glycerolipid synthesis. ACSL1 contributes more than 90% of total cardiac ACSL activity, and mice with an inducible knockout of ACSL1 (Acsl1T-/-) have impaired cardiac FA oxidation. The effects of loss of ACSL1 on mitochondrial respiratory function, phospholipid formation, or autophagic flux have not yet been studied. Acsl1T-/- hearts contained 3-fold more mitochondria with abnormal structure and displayed lower respiratory function. Because ACSL1 exhibited a strong substrate preference for linoleate (18:2), we investigated the composition of mitochondrial phospholipids. Acsl1T-/- hearts contained 83% less tetralinoleoyl-cardiolipin (CL), the major form present in control hearts. -
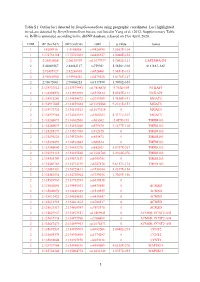
1 Table S1. Outlier Loci Detected by Deepgenomescan Using
Table S1. Outlier loci detected by DeepGenomeScan using geographic coordinates. Loci highlighted in red are detected by DeepGenomeScan but are not listed in Yang et al. (2012; Supplementary Table 4). RsID is annotated according to the dbSNP database released on 21st April, 2020. CHR BP (Grch37) BP (Grch38) rsID p.value Genes 1 1:4208918 1:4148858 rs9426495 3.8582E-104 1 1:175738168 1:175769032 rs6425357 2.0088E-105 2 2:20310668 2:20110907 rs11679737 1.2002E-111 LAPTM4A-DT 2 2:40289267 2:40062127 rs759361 5.5658E-104 SLC8A1-AS1 2 2:82495127 2:82268003 rs6726401 3.9451E-103 2 2:96660300 2:95994552 rs2579520 1.8176E-127 2 2:96672001 2:96006253 rs1917890 3.7698E-104 2 2:134333012 2:133575441 rs17816830 9.702E-105 NCKAP5 2 2:134350570 2:133592999 rs6715224 5.8745E-111 NCKAP5 2 2:134912243 2:134154672 rs2139309 1.7418E-191 MGAT5 2 2:134917005 2:134159434 rs11692586 9.2121E-151 MGAT5 2 2:134972732 2:134215161 rs11679218 0 MGAT5 2 2:134979966 2:134222395 rs1965183 5.3171E-107 MGAT5 2 2:135260071 2:134502500 rs503562 2.057E-153 TMEM163 2 2:135280039 2:134522468 rs579670 3.1477E-118 TMEM163 2 2:135285279 2:134527708 rs512375 0 TMEM163 2 2:135290221 2:134532650 rs655472 0 TMEM163 2 2:135290453 2:134532882 rs666614 0 TMEM163 2 2:135340840 2:134583270 rs842361 5.6717E-237 TMEM163 2 2:135393110 2:134635540 rs11684785 2.2164E-276 TMEM163 2 2:135430709 2:134673139 rs6745983 0 TMEM163 2 2:135469769 2:134712199 rs6747870 9.6117E-114 TMEM163 2 2:135483381 2:134725811 rs3739034 5.0357E-154 2 2:135483534 2:134725964 rs3739036 3.7269E-156 2 2:135539967 2:134782397 -

Identification of Differentially Expressed Genes in Human Bladder Cancer Through Genome-Wide Gene Expression Profiling
521-531 24/7/06 18:28 Page 521 ONCOLOGY REPORTS 16: 521-531, 2006 521 Identification of differentially expressed genes in human bladder cancer through genome-wide gene expression profiling KAZUMORI KAWAKAMI1,3, HIDEKI ENOKIDA1, TOKUSHI TACHIWADA1, TAKENARI GOTANDA1, KENGO TSUNEYOSHI1, HIROYUKI KUBO1, KENRYU NISHIYAMA1, MASAKI TAKIGUCHI2, MASAYUKI NAKAGAWA1 and NAOHIKO SEKI3 1Department of Urology, Graduate School of Medical and Dental Sciences, Kagoshima University, 8-35-1 Sakuragaoka, Kagoshima 890-8520; Departments of 2Biochemistry and Genetics, and 3Functional Genomics, Graduate School of Medicine, Chiba University, 1-8-1 Inohana, Chuo-ku, Chiba 260-8670, Japan Received February 15, 2006; Accepted April 27, 2006 Abstract. Large-scale gene expression profiling is an effective CKS2 gene not only as a potential biomarker for diagnosing, strategy for understanding the progression of bladder cancer but also for staging human BC. This is the first report (BC). The aim of this study was to identify genes that are demonstrating that CKS2 expression is strongly correlated expressed differently in the course of BC progression and to with the progression of human BC. establish new biomarkers for BC. Specimens from 21 patients with pathologically confirmed superficial (n=10) or Introduction invasive (n=11) BC and 4 normal bladder samples were studied; samples from 14 of the 21 BC samples were subjected Bladder cancer (BC) is among the 5 most common to microarray analysis. The validity of the microarray results malignancies worldwide, and the 2nd most common tumor of was verified by real-time RT-PCR. Of the 136 up-regulated the genitourinary tract and the 2nd most common cause of genes we detected, 21 were present in all 14 BCs examined death in patients with cancer of the urinary tract (1-7). -

Elucidating Biological Roles of Novel Murine Genes in Hearing Impairment in Africa
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 19 September 2019 doi:10.20944/preprints201909.0222.v1 Review Elucidating Biological Roles of Novel Murine Genes in Hearing Impairment in Africa Oluwafemi Gabriel Oluwole,1* Abdoulaye Yal 1,2, Edmond Wonkam1, Noluthando Manyisa1, Jack Morrice1, Gaston K. Mazanda1 and Ambroise Wonkam1* 1Division of Human Genetics, Department of Pathology, Faculty of Health Sciences, University of Cape Town, Observatory, Cape Town, South Africa. 2Department of Neurology, Point G Teaching Hospital, University of Sciences, Techniques and Technology, Bamako, Mali. *Correspondence to: [email protected]; [email protected] Abstract: The prevalence of congenital hearing impairment (HI) is highest in Africa. Estimates evaluated genetic causes to account for 31% of HI cases in Africa, but the identification of associated causative genes mutations have been challenging. In this study, we reviewed the potential roles, in humans, of 38 novel genes identified in a murine study. We gathered information from various genomic annotation databases and performed functional enrichment analysis using online resources i.e. genemania and g.proflier. Results revealed that 27/38 genes are express mostly in the brain, suggesting additional cognitive roles. Indeed, HERC1- R3250X had been associated with intellectual disability in a Moroccan family. A homozygous 216-bp deletion in KLC2 was found in two siblings of Egyptian descent with spastic paraplegia. Up to 27/38 murine genes have link to at least a disease, and the commonest mode of inheritance is autosomal recessive (n=8). Network analysis indicates that 20 other genes have intermediate and biological links to the novel genes, suggesting their possible roles in HI. -

An Investigation of Roles for SIRT1 and Dietary Polyphenols in Modulating the Ageing Process Through DNA Methylation
An investigation of roles for SIRT1 and dietary polyphenols in modulating the ageing process through DNA methylation Laura Jane Ions Thesis submitted for the degree of Doctor of Philosophy Institute for Cell and Molecular Biosciences, Newcastle University, UK September 2011 Declaration I certify that this thesis is my own work, except where stated, and has not been previously submitted for a degree or any other qualification at this or any other university. Laura J Ions September 2011 Thank you to Professor Dianne Ford and Dr Luisa Wakeling whose support, guidance and enthusiasm have been invaluable over the last four years. Abstract Dietary restriction (DR) can increase lifespan across evolutionarily distinct species, from yeast to rodents. The NAD+-dependent (class III) histone deacetylase SIRT1 in mammals, and its ortholog in other species, may play a major role in this response, but may affect ‘healthspan’ (number of years of good health), rather than lifespan per se. Ageing is accompanied by changes in genome methylation, which may be causal in the ageing process. Since histones are one of the many substrates that are deacetylated by SIRT1, we hypothesised that epigenetic effects of SIRT1 activity – and specifically effects on DNA methylation, which is associated closely with histone acetylation – mediate some of the beneficial effects of DR that contribute to increased healthspan. We also propose that dietary polyphenols may act at the cellular level in a similar way. To test this hypothesis, we first investigated effects of altering SIRT1 expression, by overexpression of a transgene or siRNA-mediated knockdown, on global DNA methylation (methylation of the LINE-1 element) in the human intestinal cell line, Caco-2. -

RAPGEF5 Regulates Nuclear Translocation of Β-Catenin
bioRxiv preprint doi: https://doi.org/10.1101/152892; this version posted June 20, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Title: RAPGEF5 regulates nuclear translocation of β-catenin Authors: John N. Griffin1,2, Florencia del Viso1, Anna R. Duncan1, Andrew Robson1, Saurabh Kulkarni1, Karen J. Liu2 and Mustafa K. Khokha1* Affiliations: 1 Pediatric Genomics Discovery Program, Departments of Pediatrics and Genetics Yale University School of Medicine, 333 Cedar Street, New Haven, Connecticut 06510, USA 2 Department of Craniofacial Development and Stem Cell Biology, King’s College London, London SE1 9RT, United Kingdom * Correspondence to: [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/152892; this version posted June 20, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. SUMMARY: Canonical Wnt signaling coordinates many critical aspects of embryonic development, while dysregulated Wnt signaling contributes to common diseases, including congenital malformations and cancer. The nuclear localization of β-catenin is the defining step in pathway activation. However, despite intensive investigation, the mechanisms regulating β-catenin nuclear transport remain undefined. In a patient with congenital heart disease and heterotaxy, a disorder of left-right patterning, we previously identified the guanine nucleotide exchange factor, RAPGEF5. Here, we demonstrate that RAPGEF5 regulates left-right patterning via Wnt signaling. In particular, RAPGEF5, regulates the nuclear translocation of β-catenin independently of both β-catenin cytoplasmic stabilization and the importin β1/Ran mediated transport system. -

(AMD) and Their Role in the Regulation of Gene Expression
Genetic Variants with Significant Association to Age-Related Macular Degeneration (AMD) and their Role in the Regulation of Gene Expression Dissertation zur Erlangung des Doktorgrades der Biomedizinischen Wissenschaften (Dr. rer. physiol.) der Fakultät für Medizin der Universität Regensburg vorgelegt von Tobias Strunz aus Marktredwitz im Jahr 2020 Genetic Variants with Significant Association to Age-Related Macular Degeneration (AMD) and their Role in the Regulation of Gene Expression Dissertation zur Erlangung des Doktorgrades der Biomedizinischen Wissenschaften (Dr. rer. physiol.) der Fakultät für Medizin der Universität Regensburg vorgelegt von Tobias Strunz aus Marktredwitz im Jahr 2020 Dekan: Prof. Dr. Dirk Hellwig Betreuer: Prof. Dr. Bernhard H.F. Weber Tag der mündlichen Prüfung: 02.12.2020 Parts of this work have already been published in peer-reviewed journals in an open access format: Strunz T, Grassmann F, Gayán J, Nahkuri S, Souza-Costa D, Maugeais C, Fauser S, Nogoceke E, Weber BHF (2018) A mega-analysis of expression quantitative trait loci (eQTL) provides insight into the regulatory architecture of gene expression variation in liver. Sci Rep 8: 5865. Strunz T, Lauwen S, Kiel C, den Hollander A, Weber BHF (2020) A transcriptome- wide association study based on 27 tissues identifies 106 genes potentially relevant for disease pathology in age-related macular degeneration. Sci Rep 10: 1584. Strunz T, Kiel C, Grassmann F, Ratnapriya R, Kwicklis M, Karlstetter M, Fauser S, Swaroop A, Arend N, Langmann T, Wolf A, Weber BHF (2020) A mega-analysis of expression quantitative trait loci in retinal tissue. PLoS Genet 16: e1008934. Kiel C, Berber P, Karlstetter M, Aslanidis A, Strunz T, Langmann T, Grassmann F, Weber BHF (2020) A Circulating MicroRNA Profile in a Laser-Induced Mouse Model of Choroidal Neovascularization. -

Mouse Scfd1 Knockout Project (CRISPR/Cas9)
https://www.alphaknockout.com Mouse Scfd1 Knockout Project (CRISPR/Cas9) Objective: To create a Scfd1 knockout Mouse model (C57BL/6J) by CRISPR/Cas-mediated genome engineering. Strategy summary: The Scfd1 gene (NCBI Reference Sequence: NM_029825 ; Ensembl: ENSMUSG00000020952 ) is located on Mouse chromosome 12. 25 exons are identified, with the ATG start codon in exon 1 and the TAA stop codon in exon 25 (Transcript: ENSMUST00000021335). Exon 2~5 will be selected as target site. Cas9 and gRNA will be co-injected into fertilized eggs for KO Mouse production. The pups will be genotyped by PCR followed by sequencing analysis. Note: Exon 2 starts from about 2.76% of the coding region. Exon 2~5 covers 19.51% of the coding region. The size of effective KO region: ~6324 bp. The KO region does not have any other known gene. Page 1 of 9 https://www.alphaknockout.com Overview of the Targeting Strategy Wildtype allele 5' gRNA region gRNA region 3' 1 2 3 4 5 25 Legends Exon of mouse Scfd1 Knockout region Page 2 of 9 https://www.alphaknockout.com Overview of the Dot Plot (up) Window size: 15 bp Forward Reverse Complement Sequence 12 Note: The 2000 bp section upstream of Exon 2 is aligned with itself to determine if there are tandem repeats. Tandem repeats are found in the dot plot matrix. The gRNA site is selected outside of these tandem repeats. Overview of the Dot Plot (down) Window size: 15 bp Forward Reverse Complement Sequence 12 Note: The 1754 bp section downstream of Exon 5 is aligned with itself to determine if there are tandem repeats. -

Genetics of Plgf Plasma Levels Highlights a Role of Its Receptors And
www.nature.com/scientificreports OPEN Genetics of PlGF plasma levels highlights a role of its receptors and supports the link between angiogenesis and immunity Daniela Ruggiero1,2*, Teresa Nutile1, Stefania Nappo3, Alfonsina Tirozzi2, Celine Bellenguez4, Anne‑Louise Leutenegger5,6 & Marina Ciullo1,2* Placental growth factor (PlGF) is a member of the vascular endothelial growth factor family and is involved in bone marrow‑derived cell activation, endothelial stimulation and pathological angiogenesis. High levels of PlGF have been observed in several pathological conditions especially in cancer, cardiovascular, autoimmune and infammatory diseases. Little is known about the genetics of circulating PlGF levels. Indeed, although the heritability of circulating PlGF levels is around 40%, no studies have assessed the relation between PlGF plasma levels and genetic variants at a genome‑wide level. In the current study, PlGF plasma levels were measured in a population‑based sample of 2085 adult individuals from three isolated populations of South Italy. A GWAS was performed in a discovery cohort (N = 1600), followed by a de novo replication (N = 468) from the same populations. The meta‑ analysis of the discovery and replication samples revealed one signal signifcantly associated with PlGF circulating levels. This signal was mapped to the PlGF co‑receptor coding gene NRP1, indicating its important role in modulating the PlGF plasma levels. Two additional signals, at the PlGF receptor coding gene FLT1 and RAPGEF5 gene, were identifed at a suggestive level. Pathway and TWAS analyses highlighted genes known to be involved in angiogenesis and immune response, supporting the link between these processes and PlGF regulation. Overall, these data improve our understanding of the genetic variation underlying circulating PlGF levels.