Molecular Analysis of the Diversity of Vaginal Microbiota Associated With
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chemical Structures of Some Examples of Earlier Characterized Antibiotic and Anticancer Specialized
Supplementary figure S1: Chemical structures of some examples of earlier characterized antibiotic and anticancer specialized metabolites: (A) salinilactam, (B) lactocillin, (C) streptochlorin, (D) abyssomicin C and (E) salinosporamide K. Figure S2. Heat map representing hierarchical classification of the SMGCs detected in all the metagenomes in the dataset. Table S1: The sampling locations of each of the sites in the dataset. Sample Sample Bio-project Site depth accession accession Samples Latitude Longitude Site description (m) number in SRA number in SRA AT0050m01B1-4C1 SRS598124 PRJNA193416 Atlantis II water column 50, 200, Water column AT0200m01C1-4D1 SRS598125 21°36'19.0" 38°12'09.0 700 and above the brine N "E (ATII 50, ATII 200, 1500 pool water layers AT0700m01C1-3D1 SRS598128 ATII 700, ATII 1500) AT1500m01B1-3C1 SRS598129 ATBRUCL SRS1029632 PRJNA193416 Atlantis II brine 21°36'19.0" 38°12'09.0 1996– Brine pool water ATBRLCL1-3 SRS1029579 (ATII UCL, ATII INF, N "E 2025 layers ATII LCL) ATBRINP SRS481323 PRJNA219363 ATIID-1a SRS1120041 PRJNA299097 ATIID-1b SRS1120130 ATIID-2 SRS1120133 2168 + Sea sediments Atlantis II - sediments 21°36'19.0" 38°12'09.0 ~3.5 core underlying ATII ATIID-3 SRS1120134 (ATII SDM) N "E length brine pool ATIID-4 SRS1120135 ATIID-5 SRS1120142 ATIID-6 SRS1120143 Discovery Deep brine DDBRINP SRS481325 PRJNA219363 21°17'11.0" 38°17'14.0 2026– Brine pool water N "E 2042 layers (DD INF, DD BR) DDBRINE DD-1 SRS1120158 PRJNA299097 DD-2 SRS1120203 DD-3 SRS1120205 Discovery Deep 2180 + Sea sediments sediments 21°17'11.0" -

BD™ Gardnerella Selective Agar with 5% Human Blood
INSTRUCTIONS FOR USE – READY-TO-USE PLATED MEDIA PA-254094.06 Rev.: July 2014 BD Gardnerella Selective Agar with 5% Human Blood INTENDED USE BD Gardnerella Selective Agar with 5% Human Blood is a partially selective and differential medium for the isolation of Gardnerella vaginalis from clinical specimens. PRINCIPLES AND EXPLANATION OF THE PROCEDURE Microbiological method. Gardnerella vaginalis is considered to be one of the organisms causing vaginitis.1-4 Although the organism may be present in a high percentage of normal women in the vaginal flora, its importance as a cause of non-specific vaginitis (also called bacterial vaginosis) has never been questioned. In symptomatic women, G. vaginalis frequently is associated with anaerobes such as Prevotella bivia, P. disiens, Mobiluncus, Peptostreptococcus, and/or others which are a regular part of the urethral or intestinal, but not vaginal flora. In non-specific vaginitis, normal Lactobacillus flora is reduced or absent. Gardnerella vaginalis is considered to be the indicator organism for non-specific vaginitis which, in fact, is a polymicrobial infection.3,4 Although non- culture methods such as a direct Gram stain have been recommended in recent years for genital specimens, culture is still preferred by many laboratories.1,5 G. vaginalis may also be responsible for a variety of other diseases such as preterm birth, chorioamnionitis, urinary tract infections, newborn infections, and septicemia.6 The detection of the organism on routinely used media is difficult since Gardnerella and other -

Table S5. the Information of the Bacteria Annotated in the Soil Community at Species Level
Table S5. The information of the bacteria annotated in the soil community at species level No. Phylum Class Order Family Genus Species The number of contigs Abundance(%) 1 Firmicutes Bacilli Bacillales Bacillaceae Bacillus Bacillus cereus 1749 5.145782459 2 Bacteroidetes Cytophagia Cytophagales Hymenobacteraceae Hymenobacter Hymenobacter sedentarius 1538 4.52499338 3 Gemmatimonadetes Gemmatimonadetes Gemmatimonadales Gemmatimonadaceae Gemmatirosa Gemmatirosa kalamazoonesis 1020 3.000970902 4 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas indica 797 2.344876284 5 Firmicutes Bacilli Lactobacillales Streptococcaceae Lactococcus Lactococcus piscium 542 1.594633558 6 Actinobacteria Thermoleophilia Solirubrobacterales Conexibacteraceae Conexibacter Conexibacter woesei 471 1.385742446 7 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas taxi 430 1.265115184 8 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas wittichii 388 1.141545794 9 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas sp. FARSPH 298 0.876754244 10 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sorangium cellulosum 260 0.764953367 11 Proteobacteria Deltaproteobacteria Myxococcales Polyangiaceae Sorangium Sphingomonas sp. Cra20 260 0.764953367 12 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas panacis 252 0.741416341 -

The Power of Partnership » AP2.Com MOLECULAR TEST MENU
The Power of Partnership » AP2.com MOLECULAR TEST MENU AP2 provides the most comprehensive Women’s Health Care molecular menu. AP2 can perform most of these tests from our scientifically developed UniSwabTM as well as from the liquid based platforms SurePathTM and ThinPrep®. The following is a comprehensive list of molecular tests available from AP2. If one molecular test is ordered upfront, add-on testing is available for 45 days on UniSwabTM, ThinPrep® and SurePathTM. If no molecular test is ordered up front, add-on testing is available for 28 days on UniSwabTM and ThinPrep®. HPV and CT/NG testing can be added on to ThinPrep® specimens prior to 28 days after collection. UniSwab ThinPrep SurePath 70142 Atopobium vaginae 70142 Atopobium vaginae 70142 Atopobium vaginae 60135 Bacterial vaginosis panel 60135 Bacterial vaginosis panel 60135 Bacterial vaginosis panel (Mobiluncus mulieris and M. curtisii, Gardnerella (Mobiluncus mulieris and M. curtisii, Gardnerella (Mobiluncus mulieris and M. curtisii, Gardnerella vaginalis, Atopobium vaginae, Mycoplasma vaginalis, Atopobium vaginae, Mycoplasma vaginalis, Atopobium vaginae, Mycoplasma genitalium and hominis) genitalium and hominis) genitalium and hominis) 70125 Bacteroides fragilis 70125 Bacteroides fragilis 70125 Bacteroides fragilis 70164 BVAB2 70164 BVAB2 70164 BVAB2 70551 Candida albicans 70551 Candida albicans 70551 Candida albicans 70559 Candida glabrata 70559 Candida glabrata 70559 Candida glabrata 70561 Candida kefyr 70561 Candida kefyr 70561 Candida kefyr 70560 Candida krusei 70560 -

Human Microbiota Network: Unveiling Potential Crosstalk Between the Different Microbiota Ecosystems and Their Role in Health and Disease
nutrients Review Human Microbiota Network: Unveiling Potential Crosstalk between the Different Microbiota Ecosystems and Their Role in Health and Disease Jose E. Martínez †, Augusto Vargas † , Tania Pérez-Sánchez , Ignacio J. Encío , Miriam Cabello-Olmo * and Miguel Barajas * Biochemistry Area, Department of Health Science, Public University of Navarre, 31008 Pamplona, Spain; [email protected] (J.E.M.); [email protected] (A.V.); [email protected] (T.P.-S.); [email protected] (I.J.E.) * Correspondence: [email protected] (M.C.-O.); [email protected] (M.B.) † These authors contributed equally to this work. Abstract: The human body is host to a large number of microorganisms which conform the human microbiota, that is known to play an important role in health and disease. Although most of the microorganisms that coexist with us are located in the gut, microbial cells present in other locations (like skin, respiratory tract, genitourinary tract, and the vaginal zone in women) also play a significant role regulating host health. The fact that there are different kinds of microbiota in different body areas does not mean they are independent. It is plausible that connection exist, and different studies have shown that the microbiota present in different zones of the human body has the capability of communicating through secondary metabolites. In this sense, dysbiosis in one body compartment Citation: Martínez, J.E.; Vargas, A.; may negatively affect distal areas and contribute to the development of diseases. Accordingly, it Pérez-Sánchez, T.; Encío, I.J.; could be hypothesized that the whole set of microbial cells that inhabit the human body form a Cabello-Olmo, M.; Barajas, M. -

The Characterization of a Putative Protease Expressed by Sneathia Amnii
Virginia Commonwealth University VCU Scholars Compass Theses and Dissertations Graduate School 2015 The Characterization of a Putative Protease Expressed by Sneathia amnii Rana Mehr Virginia Commonwealth University Follow this and additional works at: https://scholarscompass.vcu.edu/etd Part of the Medicine and Health Sciences Commons © The Author Downloaded from https://scholarscompass.vcu.edu/etd/3931 This Thesis is brought to you for free and open access by the Graduate School at VCU Scholars Compass. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of VCU Scholars Compass. For more information, please contact [email protected]. CHARACTERIZATION OF A PUTATIVE PROTEASE EXPRESSED BY SNEATHIA AMNII A thesis submitted in partial fulfillment of the requirements for the degree of Master of Science at Virginia Commonwealth University by RANA MEHR B.S., Virginia Commonwealth University 2011 Director: Kimberly Jefferson, Ph.D. Associate Professor, Department of Microbiology and Immunology Virginia Commonwealth University Richmond, Virginia Virginia Commonwealth University Richmond, Virginia July, 2015 Acknowledgements I would first like to express my deepest gratitude to my mentor Dr. Kimberly Jefferson. Her continuous mentorship, trust, and support in academic, scientific, and personal experiences have empowered me to successfully complete my graduate career both academically and scientifically. She has aided my development as an independent scientist which would have not been possible without guidance. I would also like to thank the members of my graduate advisory committee: Dr. Dennis Ohman and Dr. Darrell Peterson. Their advice and direction have allowed me to better understand my project and their invaluable knowledge has made me a better scientist. -
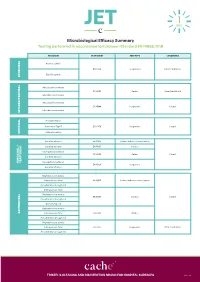
JET Microbiological Efficacy Summary
Microbiological Efficacy Summary Testing performed in accordance to European Standard EN 14885:2018 ORGANISM TEST NORM TEST TYPE CONDITIONS Bacillus subtilis EN 13704 Suspension Clean 1 and Dirty 1 Bacillus cereus SPORICIDAL Mycobacterium terrae EN 14563 Carrier Clean 1 and Dirty 2 Mycobacterium avium Mycobacterium terrae EN 14348 Suspension Clean 1 Mycobacterium avium MYCOBACTERICIDAL Poliovirus Type 1 Adenovirus Type 5 EN 14476 Suspension Clean 1 Murine Norovirus VIRUCIDAL Candida albicans EN 16615 Surface with mechanical action Candida albicans EN 13697 Surface Aspergillus brasiliensis EN 14562 Carrier Clean 1 Candida albicans YEASTICIDAL FUNGICIDAL / FUNGICIDAL Aspergillus brasiliensis EN 13624 Suspension Candida albicans Staphylococcus aureus Enterococcus hirae EN 16615 Surface with mechanical action Pseudomonas aeruginosa Enterococcus hirae Staphylococcus aureus EN 13697 Surface Clean 1 Pseudomonas aeruginosa Escherichia coli Staphylococcus aureus BACTERICIDAL Enterococcus hirae EN 14561 Carrier Pseudomonas aeruginosa Staphylococcus aureus Enterococcus hirae EN 13727 Suspension Clean 1 and Dirty 1 Pseudomonas aeruginosa TRISTEL’S CLEANING AND DISINFECTION BRAND FOR HOSPITAL SURFACES Page 1 of 3 Additional Testing TEST METHOD RNA DNA / Polyacrylamide gel electrophoresis (PAGE) ORGANISM TEST METHOD TEST TYPE CONDITIONS Acanthamoeba castellanii cysts Following the method of EN 13704 Suspension Clean 1 PROTOZOA Bacillus subtilis EN 17126 Suspension Clean 1 Bacillus cereus Clostridium difficile EN 13704 Suspension Clean 1 and Dirty 1 -

Novel Molecular, Structural and Evolutionary Characteristics of the Phosphoketolases from Bifidobacteria and Coriobacteriales
RESEARCH ARTICLE Novel molecular, structural and evolutionary characteristics of the phosphoketolases from bifidobacteria and Coriobacteriales Radhey S. Gupta*, Anish Nanda, Bijendra Khadka Department of Biochemistry and Biomedical Sciences, McMaster University, Hamilton, Ontario, Canada * [email protected] a1111111111 a1111111111 a1111111111 Abstract a1111111111 Members from the order Bifidobacteriales, which include many species exhibiting health a1111111111 promoting effects, differ from all other organisms in using a unique pathway for carbohydrate metabolism, known as the ªbifid shuntº, which utilizes the enzyme phosphoketolase (PK) to carry out the phosphorolysis of both fructose-6-phosphate (F6P) and xylulose-5-phosphate (X5P). In contrast to bifidobacteria, the PKs found in other organisms (referred to XPK) are OPEN ACCESS able to metabolize primarily X5P and show very little activity towards F6P. Presently, very lit- Citation: Gupta RS, Nanda A, Khadka B (2017) tle is known about the molecular or biochemical basis of the differences in the two forms of Novel molecular, structural and evolutionary PKs. Comparative analyses of PK sequences from different organisms reported here have characteristics of the phosphoketolases from bifidobacteria and Coriobacteriales. PLoS ONE 12 identified multiple high-specific sequence features in the forms of conserved signature (2): e0172176. doi:10.1371/journal.pone.0172176 inserts and deletions (CSIs) in the PK sequences that clearly distinguish the X5P/F6P phos- Editor: Eugene A. Permyakov, Russian Academy of phoketolases (XFPK) of bifidobacteria from the XPK homologs found in most other organ- Medical Sciences, RUSSIAN FEDERATION isms. Interestingly, most of the molecular signatures that are specific for the XFPK from Received: December 12, 2016 bifidobacteria are also shared by the PK homologs from the Coriobacteriales order of Acti- nobacteria. -

NOTES in Vitro Activities of Norfloxacin and Ciprofloxacin Against
ANTIMICROBIAL AGENTS AND CHEMOTHERAPY, July 1984, p. 94-96 Vol. 26, No. 1 0066-4804/84/070094-03$02.00/0 Copyright C 1984, American Society for Microbiology NOTES In Vitro Activities of Norfloxacin and Ciprofloxacin Against Mycobacterium tuberculosis, M. avium Complex, M. chelonei, M. fortuitum, and M. kansasii J. DOUGLAS GAY, DONALD R. DEYOUNG, AND GLENN D. ROBERTS* Section of Clinical Microbiology, Department of Laboratory Medicine, Mayo Clinic and Mayo Foundation, Rochester, Minnesota 55905 Received 28 November 1983/Accepted 4 April 1984 The activities of ciprofloxacin and norfloxacin against 100 mycobacteria isolates were studied in vitro by the 1% standard proportion method. Ciprofloxacin was more active against M. tuberculosis and M. fortuitum with MICs of 1.0 and 0.25 ,ug/ml, respectively, against 90% of isolates; norfloxacin had MICs of 8.0 and 2.0 ,ug/ml, respectively, against 90% of isolates. Nalidixic acid and other heterocyclic carbonic acid deriva- studied. The organisms were taken from the Mayo Clinic tives have been used primarily in the treatment of urinary stock culture collection, which included recent clinical iso- tract infections for many years. The compounds of this lates. Stock cultures were maintained on Middlebrook 7H10 general group include nalidixic acid, oxolinic acid, pipemidic agar slants (Difco Laboratories, Detroit, Mich.) and were acid, cinoxacin, and rosoxacin. Two new substances in this subcultured monthly. The identification of isolates was series which have been recently synthesized are norfloxacin based on standard biochemical tests (17) and gas-liquid (6) (1-ethyl-6-fluoro-1,4-dihydro-4-oxo-7-[ 1-piperazinyl ]-3- chromatography (16). -

A Genomic Journey Through a Genus of Large DNA Viruses
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Virology Papers Virology, Nebraska Center for 2013 Towards defining the chloroviruses: a genomic journey through a genus of large DNA viruses Adrien Jeanniard Aix-Marseille Université David D. Dunigan University of Nebraska-Lincoln, [email protected] James Gurnon University of Nebraska-Lincoln, [email protected] Irina V. Agarkova University of Nebraska-Lincoln, [email protected] Ming Kang University of Nebraska-Lincoln, [email protected] See next page for additional authors Follow this and additional works at: https://digitalcommons.unl.edu/virologypub Part of the Biological Phenomena, Cell Phenomena, and Immunity Commons, Cell and Developmental Biology Commons, Genetics and Genomics Commons, Infectious Disease Commons, Medical Immunology Commons, Medical Pathology Commons, and the Virology Commons Jeanniard, Adrien; Dunigan, David D.; Gurnon, James; Agarkova, Irina V.; Kang, Ming; Vitek, Jason; Duncan, Garry; McClung, O William; Larsen, Megan; Claverie, Jean-Michel; Van Etten, James L.; and Blanc, Guillaume, "Towards defining the chloroviruses: a genomic journey through a genus of large DNA viruses" (2013). Virology Papers. 245. https://digitalcommons.unl.edu/virologypub/245 This Article is brought to you for free and open access by the Virology, Nebraska Center for at DigitalCommons@University of Nebraska - Lincoln. It has been accepted for inclusion in Virology Papers by an authorized administrator of DigitalCommons@University of Nebraska - Lincoln. Authors Adrien Jeanniard, David D. Dunigan, James Gurnon, Irina V. Agarkova, Ming Kang, Jason Vitek, Garry Duncan, O William McClung, Megan Larsen, Jean-Michel Claverie, James L. Van Etten, and Guillaume Blanc This article is available at DigitalCommons@University of Nebraska - Lincoln: https://digitalcommons.unl.edu/ virologypub/245 Jeanniard, Dunigan, Gurnon, Agarkova, Kang, Vitek, Duncan, McClung, Larsen, Claverie, Van Etten & Blanc in BMC Genomics (2013) 14. -

Sneathia Species in a Case of Neonatal Meningitis from Northeast India
OMCR 20149 (3 pages) doi:10.1093/omcr/omu044 Case Report Sneathia species in a case of neonatal meningitis from Northeast India Utpala Devi1, Reeta Bora2, Jayanta Kumar Das3, Vinita Malik1 and Jagadish Mahanta1,* 1Regional Medical Research Centre, North East Region (ICMR), Dibrugarh, India, 2Neonatology Unit, Assam Medical College & Hospital, Dibrugarh, India and 3Department of Microbiology, Assam Medical College & Hospital, Dibrugarh, India Downloaded from *Correspondence address. Regional Medical Research Centre, NE Region (ICMR), Post Box 105, Dibrugarh 786001, Assam, India. Tel: þ91 373-2381494; Fax: þ91 373-2381748; E-mail: [email protected] Received 19 June 2014; revised 15 August 2014; accepted 21 August 2014 http://omcr.oxfordjournals.org/ Here we report the detection of Sneathia species most closely related to Sneathia sanguine- gens, an infrequently reported bacterium, in the cerebrospinal fluid of a neonate by a culture in- dependent method. Even though on rare occasions, this bacterium was isolated previously from the blood of neonatal bacteraemia cases. To the best of our knowledge there exists no pre- vious report of detection of S. sanguinegens in the cerebrospinal fluid even though recently there has been a report of isolation of closely related species, Leptotrichia amnionii.The neonate recovered following antimicrobial therapy for 21 days. We conclude that uncultivable or difficult- to-cultivate bacteria like Sneathia could be an emerging pathogen for neonatal at Purdue University Libraries ADMN on June 9, 2015 infection. INTRODUCTION following antimicrobial treatment in combination with pipera- cillin and netilmicin for 21 days. Sneathia is an emerging pathogen of the female genital tract having a significant role in obstetrics and gynaecological health [1]. -

Actinomyces As Actinomyces Suis Comb
INTERNATIONALJOURNAL OF SYSTEMATICBACTERIOLOGY, Jan. 1992, p. 161-165 Vol. 42, No. 1 0020-7713/92/010161-05$02.00/0 Phylogenetic Evidence for the Transfer of Eubacterium suis to the Genus Actinomyces as Actinomyces suis comb. nov. W. LUDWIG,l* G. KIRCHHOF,l M. WEIZENEGGER,l AND N. WEISS2 Lehrstuhl fur Mikrobiologie, Technische Universitat, 0-8000 Munich, and Deutsche Sammlung von Mikroorganismen und Zellkulturen, 0-3300 Braunschweig, Germany The 16s rRNA primary structures of Eubacterium suis DSM 20639T (T = type strain) and Bijidobacterium bijidum DSM 20456T were determined by sequencing in vitro amplified rDNA. Sequence comparisons indicated that B. bijidum is moderately related to representatives of the genera Actinomyces and Mobiluncus. The closest relative of E. suis is Actinomyces pyogenes. E. suis and A. pyogenes are more closely related phylogenetically to one another than to the other Actinomyces species that have been investigated by using comparative 16s rRNA analysis. Therefore, we propose that E. suis should be transferred to the genus Actinomyces as Actinomyces suis comb. nov. Eubacterium suis Wegienek and Reddy 1982, a commonly MATERIALS AND METHODS occurring swine pathogen (23), was originally isolated by Soltys and Spratling in 1957 (20). The name “Corynebacte- Bacterial strains and culture conditions. B. bifidum DSM rium suis” was proposed for this bacterium because of the 20456T (T = type strain) was cultured anaerobically at 37°C diphtheroid morphology of the anaerobic organism. In 1982 in a medium which contained (per liter of distilled water) 10 in a taxonomic study, Wegienek and Reddy described the g of tryptone, 5 g of meat extract, 5 g of yeast extract, 10 g morphological, cultural, and biochemical characteristics of of glucose, 3 g of K2HP0,, 5 g of NaC1, 10 ml of Tween 80, this organism and proposed the name Eubacterium suis.