Surfen, a Small Molecule Antagonist of Heparan Sulfate
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A 74-Year-Old Woman with Abdominal Pain and Fever
A SELF-TEST IM BOARD REVIEW DAVID L. LONGWORTH, MO, JAMES K. STOLLER, MD, EDITORS OF CLINICAL PETER MAZZONE, MD CRAIG NIELSEN, MD RECOGNITION Department of Pulmonary and Critical Department of General Internal Care Medicine, Cleveland Clinic Medicine, Cleveland Clinic A 74-year-old woman with abdominal pain and fever 74-YEAR-OLD WOMAN was transferred pulmonary embolism, and a subsequent pul- from a local hospital for further evalua- monary angiogram was read as normal. A tion and management of abdominal pain with chest CT scan did not reveal anything other fever. than the small bilateral pleural effusions seen The patient had presented to the local on the chest radiograph. A thoracentesis hospital 11 days before with a 1-day history of revealed transudative fluid only. Intravenous bilateral upper-quadrant abdominal pain. She heparin was discontinued. described the pain as a constant ache with Past history. The patient had had essen- intermittent sharper pains accompanied by tial thrombocythemia for 5 years, for which nausea, but she could not identify any precip- she took hydroxyurea until 2 months before itants of the pain. She was also constipated. admission. Hydroxyurea was restarted at the A few days after being admitted to the local hospital because her platelet count was local hospital, the patient had developed a high at 750 x 109/L (normal 150-400 x fever and mild shortness of breath. She was 109/L), but it was stopped 3 days later because treated empirically for a possible pulmonary of mucositis. More than 30 years ago, the infection with a variety of antibiotics (ceftri- patient had been diagnosed with "pernicious Her platelet axone, ticarcillin-clavulanate, erythromycin, anemia. -

Dermatan Sulfate Composition
Europäisches Patentamt *EP000671414B1* (19) European Patent Office Office européen des brevets (11) EP 0 671 414 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.7: C08B 37/00, A61K 31/715 of the grant of the patent: 02.01.2002 Bulletin 2002/01 (86) International application number: PCT/JP94/01643 (21) Application number: 94927827.9 (87) International publication number: (22) Date of filing: 30.09.1994 WO 95/09188 (06.04.1995 Gazette 1995/15) (54) DERMATAN SULFATE COMPOSITION AS ANTITHROMBOTIC DERMATANSULFATEZUSAMMENSETZUNG ALS ANTITHROMOTISCHES MITTEL COMPOSITION DE DERMATANE SULFATE UTILISE COMME ANTITHROMBOTIQUE (84) Designated Contracting States: • YOSHIDA, Keiichi AT BE CH DE DK ES FR GB IT LI NL PT SE Tokyo 189 (JP) (30) Priority: 30.09.1993 JP 26975893 (74) Representative: Davies, Jonathan Mark Reddie & Grose 16 Theobalds Road (43) Date of publication of application: London WC1X 8PL (GB) 13.09.1995 Bulletin 1995/37 (56) References cited: (60) Divisional application: EP-A- 0 112 122 EP-A- 0 199 033 01201321.5 / 1 125 977 EP-A- 0 238 994 WO-A-93/05074 JP-A- 59 138 202 JP-B- 2 100 241 (73) Proprietor: SEIKAGAKU KOGYO KABUSHIKI JP-B- 6 009 042 KAISHA Tokyo 103 (JP) • CARBOHYDRATE RESEARCH , vol. 131, no. 2, 1984 NL, pages 301-314, K. NAGASAWA ET AL. (72) Inventors: ’The structure of rooster-comb dermatan • TAKADA, Akikazu sulfate. Characterization and quantitative Shizuoka 431-31 (JP) determination of copolymeric, isomeric tetra- • ONAYA, Junichi and hexa-saccharides’ Higashiyamato-shi Tokyo 207 (JP) • ARAI, Mikio Remarks: Higashiyamato-shi Tokyo 207 (JP) The file contains technical information submitted • MIYAUCHI, Satoshi after the application was filed and not included in this Tokyo 208 (JP) specification • KYOGASHIMA, Mamoru Tokyo 207 (JP) Note: Within nine months from the publication of the mention of the grant of the European patent, any person may give notice to the European Patent Office of opposition to the European patent granted. -

Avoiding Intelligence Failures in the Cardiac Catheterization Laboratory
Review Avoiding Intelligence Failures in the Cardiac Catheterization Laboratory: Strategies for the Safe and Rational Use of Dalteparin or Enoxaparin during Percutaneous Coronary Intervention Jonathan D. Marmur, MD, Renee P. Bullock-Palmer, MD, Shyam Poludasu, MD, Erdal Cavusoglu, MD ABSTRACT: Low-molecular-weight heparin (LMWH) has been a of unstable angina (UA) and non-ST-segment elevation myocardial mainstay for the management of acute coronary syndromes (ACS) for infarction (NSTEMI). The ESSENCE and TIMI 11B trials support almost a decade. However, several recent developments have seriously the use of LMWH over unfractionated heparin (UFH) in acute coro - threatened the prominence of this drug class: (i) the adoption of an nary syndromes managed with a predominantly medical approach early invasive strategy, frequently leading to percutaneous coronary in - 1,2 tervention (PCI) where the dosing and monitoring of LMWH is un - as opposed to an invasive initial strategy. familiar to most operators, (ii) the results of the SYNERGY trial, which Since the publication of the ESSENCE and TIMI 11B trials, not only failed to establish the superiority of enoxaparin over unfrac - the management of acute coronary syndromes (ACS) has tionated heparin with respect to efficacy, but also demonstrated more evolved to favor an early invasive strategy. This evolution is sup - bleeding with LMWH, and (iii) the results of the REPLACE-2 and ported by a number of studies, including the FRISC II and the ACUITY trials, which have demonstrated the advantages of an ACS TACTICS TIMI-18 trials. 3,4 The Superior Yield of the New and PCI treatment strategy based on direct thrombin inhibition with bivalirudin. -
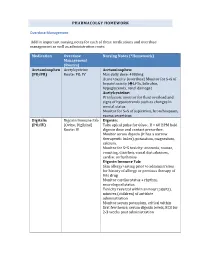
PHARMACOLGY HOMEWORK Overdose Management Add In
PHARMACOLGY HOMEWORK Overdose Management Add in important nursing notes for each of these medications and overdose management as well as administration route. Medication Overdose Nursing Notes (*Homework) Management (Routes) Acetaminophen Acetylcysteine Acetaminophen: (PO/PR) Route: PO, IV Max daily dose: 4000mg Acute toxicity (overdose) Monitor for S+S of hepatotoxicity (éLFTs, bilirubin, hypoglycemia, renal damage) Acetylcysteine: IV infusion: monitor for fluid overload and signs of hyponatremia such as changes in mental status Monitor for S+S of aspiration, bronchospasm, excess secretions Digitalis Digoxin Immune Fab Digoxin: (PO/IV) (Ovine, Digibind) Take apical pulse for 60sec. If < 60 BPM hold Route: IV digoxin dose and contact prescriber. Monitor serum digoxin (it has a narrow therapeutic index), potassium, magnesium, calcium. Monitor for S+S toxicity: anorexia, nausea, vomiting, diarrhea, visual disturbances, cardiac arrhythmias Digoxin Immune Fab: Skin allergy testing prior to administration for history of allergy or previous therapy of this drug Monitor cardiac status + rhythm, neurological status Toxicity reversal within an hour (adults), minutes (children) of antidote administration Monitor serum potassium, critical within first few hours; serum digoxin levels, ECG for 2-3 weeks post administration Heparin Protamine sulfate Heparin: (IV/SC) (IV) Monitor for spontaneous bleeding, thrombocytopenia Monitor aPTT levels Protamine Sulfate: Sudden drop in BP Monitor BP+ P q15-30 min Monitor aPTT Opioid Naloxone (IV-adults, Opioids: -

Jp Xvii the Japanese Pharmacopoeia
JP XVII THE JAPANESE PHARMACOPOEIA SEVENTEENTH EDITION Official from April 1, 2016 English Version THE MINISTRY OF HEALTH, LABOUR AND WELFARE Notice: This English Version of the Japanese Pharmacopoeia is published for the convenience of users unfamiliar with the Japanese language. When and if any discrepancy arises between the Japanese original and its English translation, the former is authentic. The Ministry of Health, Labour and Welfare Ministerial Notification No. 64 Pursuant to Paragraph 1, Article 41 of the Law on Securing Quality, Efficacy and Safety of Products including Pharmaceuticals and Medical Devices (Law No. 145, 1960), the Japanese Pharmacopoeia (Ministerial Notification No. 65, 2011), which has been established as follows*, shall be applied on April 1, 2016. However, in the case of drugs which are listed in the Pharmacopoeia (hereinafter referred to as ``previ- ous Pharmacopoeia'') [limited to those listed in the Japanese Pharmacopoeia whose standards are changed in accordance with this notification (hereinafter referred to as ``new Pharmacopoeia'')] and have been approved as of April 1, 2016 as prescribed under Paragraph 1, Article 14 of the same law [including drugs the Minister of Health, Labour and Welfare specifies (the Ministry of Health and Welfare Ministerial Notification No. 104, 1994) as of March 31, 2016 as those exempted from marketing approval pursuant to Paragraph 1, Article 14 of the Same Law (hereinafter referred to as ``drugs exempted from approval'')], the Name and Standards established in the previous Pharmacopoeia (limited to part of the Name and Standards for the drugs concerned) may be accepted to conform to the Name and Standards established in the new Pharmacopoeia before and on September 30, 2017. -

Low Molecular Weight Heparinoid, ORG 10172 (Danaparoid), and Outcome After Acute Ischemic Stroke a Randomized Controlled Trial
Original Contributions Low Molecular Weight Heparinoid, ORG 10172 (Danaparoid), and Outcome After Acute Ischemic Stroke A Randomized Controlled Trial The Publications Committee for the Trial of ORG 10172 in Acute Stroke Treatment (TOAST) Investigators Context.—Anticoagulation with unfractionated heparin is used commonly for ANTICOAGULATION with unfrac- treatment of acute ischemic stroke, but its use remains controversial because it has tionated heparin commonly is used to not been shown to be effective or safe. Low molecular weight heparins and hepa- treat persons with acute ischemic 1 rinoids have been shown to be effective in preventing deep vein thrombosis in per- stroke. However, the use of heparin re- mains controversial because it is not es- sons with stroke, and they might be effective in reducing unfavorable outcomes fol- 2-6 lowing ischemic stroke. tablished as safe or effective. A recent open trial demonstrated a modest effect Objective.—To test whether an intravenously administered low molecular from subcutaneously administered hep- weight heparinoid, ORG 10172 (danaparoid sodium), increases the likelihood of a arin in preventing recurrent stroke favorable outcome at 3 months after acute ischemic stroke. within 14 days but no improvement in Design.—Randomized, double-blind, placebo-controlled, multicenter trial. outcomes.7 Thus, whether an intrave- Setting and Participants.—Between December 22, 1990, and December 6, nously administered anticoagulant that 1997, 1281 persons with acute stroke were enrolled at 36 centers across the United would act more rapidly would be effec- States. tiveremainsunanswered.Thesearchfor Intervention.—A 7-day course of ORG 10172 or placebo was given initially as alternative medications that possess the a bolus within 24 hours of stroke, followed by continuous infusion in addition to the antithrombotic characteristics of hepa- best medical care. -

Estonian Statistics on Medicines 2016 1/41
Estonian Statistics on Medicines 2016 ATC code ATC group / Active substance (rout of admin.) Quantity sold Unit DDD Unit DDD/1000/ day A ALIMENTARY TRACT AND METABOLISM 167,8985 A01 STOMATOLOGICAL PREPARATIONS 0,0738 A01A STOMATOLOGICAL PREPARATIONS 0,0738 A01AB Antiinfectives and antiseptics for local oral treatment 0,0738 A01AB09 Miconazole (O) 7088 g 0,2 g 0,0738 A01AB12 Hexetidine (O) 1951200 ml A01AB81 Neomycin+ Benzocaine (dental) 30200 pieces A01AB82 Demeclocycline+ Triamcinolone (dental) 680 g A01AC Corticosteroids for local oral treatment A01AC81 Dexamethasone+ Thymol (dental) 3094 ml A01AD Other agents for local oral treatment A01AD80 Lidocaine+ Cetylpyridinium chloride (gingival) 227150 g A01AD81 Lidocaine+ Cetrimide (O) 30900 g A01AD82 Choline salicylate (O) 864720 pieces A01AD83 Lidocaine+ Chamomille extract (O) 370080 g A01AD90 Lidocaine+ Paraformaldehyde (dental) 405 g A02 DRUGS FOR ACID RELATED DISORDERS 47,1312 A02A ANTACIDS 1,0133 Combinations and complexes of aluminium, calcium and A02AD 1,0133 magnesium compounds A02AD81 Aluminium hydroxide+ Magnesium hydroxide (O) 811120 pieces 10 pieces 0,1689 A02AD81 Aluminium hydroxide+ Magnesium hydroxide (O) 3101974 ml 50 ml 0,1292 A02AD83 Calcium carbonate+ Magnesium carbonate (O) 3434232 pieces 10 pieces 0,7152 DRUGS FOR PEPTIC ULCER AND GASTRO- A02B 46,1179 OESOPHAGEAL REFLUX DISEASE (GORD) A02BA H2-receptor antagonists 2,3855 A02BA02 Ranitidine (O) 340327,5 g 0,3 g 2,3624 A02BA02 Ranitidine (P) 3318,25 g 0,3 g 0,0230 A02BC Proton pump inhibitors 43,7324 A02BC01 Omeprazole -

Search Strategy, Baseline Risk Coronavirus and Prophylaxis For
Supplement 7: Search Strategy, Baseline Risk Coronavirus and prophylaxis for thrombotic events Search narrative, 23 July 2020 Bibliographic databases: EMBASE (1974 to, 22 July 2020) Epistemonikos COVID-19 Evidence (21 July 2020) (includes records from Cochrane Database of Systematic Reviews, Pubmed, EMBASE, CINAHL, PsycINFO, LILACS, Database of reviews of Effects, The Campbell Collaboration online library, JBI Database of Systematic Reviews and Implementation Reports, EPPI-Centre Evidence Library) MEDLINE (1946 to, 23 July 2020) SCOPUS (19 July 2020) WHO Global Research Database (COVID-19) (20 July 2020) Trial/study Databases: Cochrane COVID-19 study register (23 July 2020) (includes records from ClinicalTrials.gov, WHO ICTRN, and PubMed) CYTEL map of ongoing [COVID-19] clinical trials (20 July 2020) (WHO International Clinical Trials Registry Platform, European Clinical Trials Registry, clinicaltrials.gov, Chinese Clinical Trial Registry, German Clinical Trials registry, Japan Primary Registries Network, Iranian Clinical Trial Registry, and Australian New Zealand Clinical Trials Registry) Results were downloaded from sources and deduplicated in EndNote. Results from BioRxiV, ChemRxiV, MedRxiV (preprints indexed in Medline and Epistemonikos), or protocols for clinical trials were removed as they were beyond the scope of the search. COCHRANE COVID-19 (https://covid-19.cochrane.org/) Search 1: anticoagula* or anti-coagula* or coagulation or thromb* or antithromb* or antiplatelet* or heparin or LMWH or aspirin or embol* or phlebothrombos* -

Antidote List
Antidote List Recommended minimum amounts for hospital pharmacies to stock Call the Maryland Poison Center for expert advice on the treatment of all poisonings and overdoses: 1-800-222-1222 Poisoning Minimum Stocking Antidote Indication Recommendations Oral: 120 grams Acetylcysteine Acetaminophen IV: 96 grams Crotaline snake Antivenin, snake (CroFab®) 12 vials envenomation 1 vial Black widow spider (Note: Merck limits distribution Antivenin, black widow spider envenomation to cases of confirmed bites with symptoms only) Organophosphate & Atropine carbamate insecticides, nerve 165 mg gases Calcium chloride Calcium channel blockers 10 grams 2.25 grams Calcium disodium EDTA Lead (Available direct from ASD Specialty Healthcare) Hydrofluoric acid 1 kg (powder) Calcium gluconate Calcium channel blockers IV: 30 grams 1 kit Cyanide Antidote Kit Cyanide (or Hydroxocobalamin, see below) Cyproheptadine (Periactin®) Serotonin syndrome 80 mg Neuroleptic malignant Dantrolene syndrome, stimulant-induced 720 mg hyperthermia Deferoxamine mesylate Iron 8 grams (Desferal®) Digoxin Immune FAB Digoxin 20 vials (Digibind®, DigiFab®) Dimercaprol (BAL) Heavy metals 1.5 grams 1 | P a g e Maryland Poison Center Antidote List – continued Poisoning Minimum Stocking Antidote Indication Recommendations DMSA (Succimer, Chemet®) Heavy metals 2000 mg Folic acid Methanol IV: 150 mg Flumazenil (Romazicon®) Benzodiazepines 10 mg Fomepizole (Antizol®) Ethylene glycol, methanol 12 grams Beta blockers, Glucagon 50 mg calcium channel blockers Hydroxocobalamin (Cyanokit®) Cyanide -

Products for Heparin Analysis
Products for Heparin Analysis 2014 - 2015 Adhoc International Our Company Located in Beijing, Adhoc International is an integrated vendor who produces reagents used in researches and quality control of heparins and chondroitin sulfates. Adhoc International also uses its engineering prowess to develop novel devices for microbial mutation, such as multifunctional plasma mutagenesis systems. Our Focuses Heparinases and chondroitinases Reagents for chromogenic assays Determination of heparin sources Multifunctional plasma mutagenesis systems Chemicals used in personal health products Table of Contents Heparinases 2 Chondroitinases 4 Chromogenic Assays 6 GAG Disaccharides 8 Heparin Analogs 10 Heparin Polysaccharides 11 Benzethonium Chloride 12 1 www.aglyco.com Heparinases from Flavobacterium heparinum Heparinases, or heparin lyases, are widely used in studies of heparin and heparan sulfate as well as in quality control of heparin products. With improved fermentation capabilities and advanced purification techniques, we promise a large supply of natural heparinases. Specificity Heparinase I & II Heparinase II & III D-Glucosamine D-Glucosamine L-Iduronic acid D-Glucuronic acid N-Acetyl-D-glucosamine Heparinases can clave glycosidic bonds of heparin and/or heparan sulfate by a β-elimination mechanism, generating unsaturated products (mostly disaccharides) with a double bond be- tween C4 and C5 of the uronate residue. The resulting unsaturated products can be measured at 232 nm wavelength. Enzyme Substrate Heparinase I Heparin and heparan -

Dermatan Sulfate Is the Predominant Antithrombotic Glycosaminoglycan in Vessel Walls: Implications for a Possible Physiological Function of Heparin Cofactor II
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Biochimica et Biophysica Acta 1740 (2005) 45–53 http://www.elsevier.com/locate/bba Dermatan sulfate is the predominant antithrombotic glycosaminoglycan in vessel walls: Implications for a possible physiological function of heparin cofactor II Ana M.F. Tovar, Diogo A. de Mattos, Mariana P. Stelling, Branca S.L. Sarcinelli-Luz, Roˆmulo A. Nazareth, Paulo A.S. Moura˜o* Laborato´rio de Tecido Conjuntivo, Hospital Universita´rio Clementino Fraga Filho and Instituto de Bioquı´mica Me´dica, Centro de Cieˆncias da Sau´de, Universidade Federal do Rio de Janeiro, Caixa Postal 68041, Rio de Janeiro, RJ, 21941-590, Brazil Received 10 December 2004; received in revised form 17 February 2005; accepted 23 February 2005 Available online 11 March 2005 Abstract The role of different glycosaminoglycan species from the vessel walls as physiological antithrombotic agents remains controversial. To further investigate this aspect we extracted glycosaminoglycans from human thoracic aorta and saphenous vein. The different species were highly purified and their anticoagulant and antithrombotic activities tested by in vitro and in vivo assays. We observed that dermatan sulfate is the major anticoagulant and antithrombotic among the vessel wall glycosaminoglycans while the bulk of heparan sulfate is a poorly sulfated glycosaminoglycan, devoid of anticoagulant and antithrombotic activities. Minor amounts of particular a heparan sulfate (b5% of the total arterial glycosaminoglycans) with high anticoagulant activity were also observed, as assessed by its retention on an antithrombin-affinity column. Possibly, this anticoagulant heparan sulfate originates from the endothelial cells and may exert a significant physiological role due to its location in the interface between the vessel wall and the blood. -

Protamine Sulfate Enhances Lipid-Mediated Gene Transfer
Gene Therapy (1997) 4, 961–968 1997 Stockton Press All rights reserved 0969-7128/97 $12.00 Protamine sulfate enhances lipid-mediated gene transfer FL Sorgi1,2, S Bhattacharya1 and L Huang1 1The Laboratory of Drug Targeting, Department of Pharmacology, The University of Pittsburgh School of Medicine, Pittsburgh, PA, USA A polycationic peptide, protamine sulfate, USP, has been USP as a condensation agent was found to be superior to shown to be able to condense plasmid DNA efficiently for poly-L-lysine as well as to various other types of protamine. delivery into several different types of cells in vitro by sev- These differences among various salt forms of protamine eral different types of cationic liposomes. The monovalent appear to be attributable to structural differences between cationic liposomal formulations (DC-Chol and lipofectin) the protamines and not due to differences in the net charge exhibited increased transfection activities comparable to of the molecule. The appearance of lysine residues within that seen with the multivalent cationic liposome formu- the protamine molecule correlate with a reduction in bind- lation, lipofectamine. This suggests that lipofectamine’s ing affinity to plasmid DNA, as well as an observed loss in superior in vitro activity arises from its ability to condense transfection-enhancing activity. This finding sheds light on DNA efficiently and that protamine’s primary role is that of the structural requirements of condensation agents for use a condensation agent, although it also possesses several in gene transfer protocols. Furthermore, protamine sulfate, amino acid sequences resembling that of a nuclear localiz- USP is an FDA-approved compound with a documented ation signal.