Organocopper Compounds
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

And C,N-Chelated Organocopper Compounds†
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 2 September 2021 Review C,C- and C,N-Chelated Organocopper Compounds† Liang Liu1, Hui Chen2, Zhenqiang Yang2, Junnian Wei1* and Zhenfeng Xi1,3* 1 Beijing National Laboratory for Molecular Sciences (BNLMS), Key Laboratory of Bioorganic Chemistry and Molecular Engineering of Ministry of Education, College of Chemistry, Peking University, Beijing 100871, China; [email protected] (L.L.), [email protected] (J.W.), [email protected] (Z.X.) 2 Henan Institute of Chemistry Co. Ltd., Henan Academy of Sciences, Zhengzhou 450002, China; [email protected] (H.C.), [email protected] (Z.Y.) 3 State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry (SIOC), Shanghai 200032, China; [email protected] (Z.X.) * Correspondence: [email protected] (J.W.), [email protected] (Z.X.) † Dedicated to Professor Gerard van Koten on the occasion of his 80th birthday Abstract: Copper-catalyzed and organocopper-involved reactions are of great significance in organic synthesis. To have a deep understanding of the reaction mechanisms, the structural characterizations of organocopper intermediates become indispensable. Meanwhile, the structure- function relationship of organocopper compounds would advance rational design and development of new Cu-based reactions and organocopper reagents. Compared to the mono- carbonic ligand, the C,N- and C,C-bidentate ligands better stabilize the unstable organocopper compounds. The bidentate ligands can chelate to the same copper atom via 휂2-mode, forming a mono-cupra-cyclic compounds with at least one acute C-Cu-C angle. When the bidentate ligands bind to two copper atoms via 휂1-mode at each coordinating site, the bimetallic macrocyclic compounds will form nearly linear C-Cu-C angles. -

Antifouling Coating Composition, Coating Film Therefrom, Underwater Material Covered with the Coating Film and Antifouling Method
Europäisches Patentamt *EP001342756A1* (19) European Patent Office Office européen des brevets (11) EP 1 342 756 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.7: C09D 5/16 10.09.2003 Bulletin 2003/37 (21) Application number: 03251373.1 (22) Date of filing: 06.03.2003 (84) Designated Contracting States: • Nakamura, Naoya AT BE BG CH CY CZ DE DK EE ES FI FR GB GR Ohtake-shi, Hiroshima (JP) HU IE IT LI LU MC NL PT RO SE SI SK TR • Tsuboi, Makoto Designated Extension States: Ohtake-shi, Hiroshima (JP) AL LT LV MK (74) Representative: Cresswell, Thomas Anthony (30) Priority: 06.03.2002 JP 2002060696 J.A. KEMP & CO. 14 South Square (71) Applicant: CHUGOKU MARINE PAINTS, LTD. Gray’s Inn Ohtake-shi, Hiroshima (JP) London WC1R 5JJ (GB) (72) Inventors: • Oya, Masaaki, Ohtake-shi, Hiroshima (JP) (54) Antifouling coating composition, coating film therefrom, underwater material covered with the coating film and antifouling method (57) An antifouling coating composition comprising: (D) a dehydrating agent. (A) a silyl ester copolymer containing constituent units derived from a polymerizable unsaturated car- boxylic acid silyl ester, (B) a carboxylic acid, (C) a bivalent or trivalent metal compound, and EP 1 342 756 A1 Printed by Jouve, 75001 PARIS (FR) EP 1 342 756 A1 Description FIELD OF THE INVENTION 5 [0001] The present invention relates to an antifouling coating composition which contains a silyl ester copolymer, an antifouling coating film formed from the antifouling coating composition, an antifouling method wherein the antifouling coating composition is used, and a marine vessel (hull) or underwater structure covered with the coating film. -
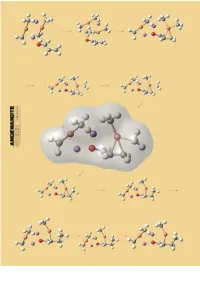
Structures and Reaction Mechanisms of Organocuprate Clusters in Organic Chemistry
REVIEWS Wherefore Art Thou Copper? Structures and Reaction Mechanisms of Organocuprate Clusters in Organic Chemistry Eiichi Nakamura* and Seiji Mori Organocopper reagents provide the principles. This review will summarize example of molecular recognition and most general synthetic tools in organic first the general structural features of supramolecular chemistry, which chemistry for nucleophilic delivery of organocopper compounds and the pre- chemists have long exploited without hard carbanions to electrophilic car- vious mechanistic arguments, and then knowing it. Reasoning about the bon centers. A number of structural describe the most recent mechanistic uniqueness of the copper atom among and mechanistic studies have been pictures obtained through high-level neighboring metal elements in the reported and have led to a wide variety quantum mechanical calculations for periodic table will be presented. of mechanistic proposals, some of three typical organocuprate reactions, which might even be contradictory to carbocupration, conjugate addition, Keywords: catalysis ´ conjugate addi- others. With the recent advent of and SN2 alkylation. The unified view tions ´ copper ´ density functional physical and theoretical methodolo- on the nucleophilic reactivities of met- calculations ´ supramolecular chemis- gies, the accumulated knowledge on al organocuprate clusters thus ob- try organocopper chemistry is being put tained has indicated that organocup- together into a few major mechanistic rate chemistry represents an intricate 1. Introduction 1 R Cu X The desire to learn about the nature of elements has been R or R1 R and will remain a main concern of chemists. In this review, we R Cu will consider what properties of copper make organocopper R1 chemistry so useful in organic chemistry. -

Studies Towards a Total Synthesis of Hippeastrine Johanna Myriam Helary 4769139
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by University of East Anglia digital repository Studies towards a total synthesis of Hippeastrine Johanna Myriam Helary 4769139 © I certify that the work contained in the thesis submitted by me for the degree of Master of Science by Research is my original work except where due reference is made to other authors, and has not been previously submitted by me for a degree at this or any other university. 1 Acknowledgements I would like to thanks my mum, my stepdad and Deeptee Horil Roy for their continued supports and encouragements over the last five years. For that I am very grateful. I would also like to thank all of the members Dr G. R Stephenson and Dr C. Richard with special thanks to Dr Julien Doulcet, Dr Paulina Glowacka and the rest of Floor 3 for their help, advices, patient explanations, and good humour over the last two years. Finally I would like to thank Dr. G. R Stephenson for all of his support, guidance and encouragements. In the loving memory of Sarah Delf, devoted friend and colleague. Wherever you are you always will be remembered. Rest in peace my amazing friend and fellow Hippeastrine crew member. 2 Table of Contents Abstract ................................................................................................................................. 5 1. Introduction .......................................................................................................................... 6 1.1. History of alkaloids ....................................................................................................... -

Hoto P QH PH
US010211400B2 (12 ) United States Patent ( 10 ) Patent No. : US 10 ,211 , 400 B2 Hawker et al. ( 45 ) Date of Patent: Feb . 19 , 2019 (54 ) PHOTOPATTERNED GROWTH OF (51 ) Int . Ci. ELECTRONICALLY ACTIVE BRUSH HOIL 51/ 00 ( 2006 . 01 ) POLYMERS FOR LIGHT EMITTING DIODE C09K 11 / 06 (2006 . 01) DISPLAYS (Continued ) (71 ) Applicants : The Regents of the University of ( 52 ) U .S . CI. California , Oakland , CA (US ) ; Rohm CPC . .. .. HOIL 51 /0035 (2013 . 01 ) ; C09K 11/ 06 and Haas Electronic Materials LLC , ( 2013. 01 ) ; HOIL 51/ 0043 ( 2013 .01 ) ; Marlborough , MA (US ) ; Dow Global ( Continued ) Technologies LLC , Midland , MI (US ) (58 ) Field of Classification Search CPC .. .. .. HO1L 51 /0035 ; HO1L 51 /0043 ; HOIL ( 72 ) Inventors : Craig J . Hawker , Santa Barbara , CA (58 ) Fieldof c . 51. .HOZZ /5072 ; HOLLHOIL 551 / 5088 ; HO1L 51/ 5206 ; (US ) ; Zachariah Allen Page , Goleta , CA (US ) ; Peter Trefonas, III, Medway , (Continued ) MA (US ) ; Anatoliy N . Sokolov , Midland , MI (US ) ; John Kramer , ( 56 ) References Cited Midland , MI (US ) ; David S . Laitar, U . S . PATENT DOCUMENTS Midland , MI (US ) ; Sukrit Mukhopadhyay , Midland , MI (US ) ; 6 ,423 , 465 B1 7 /2002 Hawker et al . Benjaporn Narupai, Goleta , CA (US ) ; 6 , 447 , 879 B19 / 2002 Sakurai et al . Christian Wilhelm Pester , Goleta , CA ( Continued ) (US ) OTHER PUBLICATIONS (73 ) Assignees : DOW GLOBAL TECHNOLOGIES LLC , Midland , MI (US ) ; ROHM AND Choi et al. ; “ Simple Detachment Patterning of Organic Layers and HAAS ELECTRONIC MATERIALS Its Application to Organic Light- Emitting Diodes ” ; Adv. Mater. ; 17 , LLC , Marlborough , MA (US ) ; THE No . 2 ; Jan . 31 , 2005 , pp . 166 - 171 . REGENTS OF THE UNIVERSITY (Continued ) OF CALIFORNIA , Oakland, CA (US ) Primary Examiner — Su C Kim ( * ) Notice: Subject to any disclaimer, the term of this ( 74 ) Attorney , Agent , or Firm — Cantor Colburn LLP patent is extended or adjusted under 35 U . -

Decarboxylative Cross-Coupling: Bimetallic Nanoparticles As Catalysts and Low-Temperature Optimisation
Decarboxylative Cross-coupling: Bimetallic Nanoparticles as Catalysts and Low-Temperature Optimisation Joseph Kyle Sheppard Submitted in accordance with the requirements for the degree of Doctor of Philosophy University of Leeds School of Chemistry September 2020 The candidate confirms the work submitted is his own and that appropriate credit has been given where reference has been made to the work of others. I Acknowledgements First and foremost, I would like to thank my supervisors Dr. Bao N. Nguyen and Prof. Patrick McGowan for providing me with the opportunity to undertake this PhD. Bao, you deserve a medal for putting up with me for four whole years! Thank you dearly for everything you have taught me. Patrick, though our meetings were few and far between, I always left them brimming with new ideas and knowledge. I must also thank the wonderful Dr. Alexander Kulak; chiefly for all of your assistance with AAS and SEM analysis but also for being a friendly face that never failed to brighten my days. I’ll deeply miss the hours we spent, shouting over the noise of the spectrometer and trying to work out why it would develop an entirely new issue every time we used it. I would also like to thank Dr. Rebecca Stones for all of her hard work running my TEM samples and teaching me how to analyse the micrographs. Special thanks must also go to Dr. Mary Bayana for her patience in teaching me how to use all of the arcane and intricate analytical machines of the iPRD and to Martin Huscroft for helping me refine my HPLC method. -

Synthetic Organic Chemistry Mscch-07 Vardhman Mahaveer Open University, Kota
MScCH-07 Vardhman Mahaveer Open University, Kota Synthetic Organic Chemistry MScCH-07 Vardhman Mahaveer Open University, Kota Synthetic Organic Chemistry Course Development Committee Chair Person Prof. Ashok Sharma Vice-Chancellor Vardhman Mahaveer Open University, Kota Coordinator and Members Coordinator SANDEEP HOODA Assistant Professor of Zoology School of Science & Technology Vardhman Mahaveer Open University, Kota Prof L.R. Gurjar Dr. Arvind Pareek Director Academic Director Regional Centre-Bharatpur VMOU Kota VMOU Kota Dr. Anuradha Dubey Dr. Sunil kumar Jangir Deputy Director,SOST Convener Chemistry VMOU Kota VMOU Kota Prof. P.S. Verma (Retd.) Prof. Pahup Singh (Retd.) Department of Chemistry Department of Chemistry University of Raj, Jaipur University of Raj, Jaipur Prof. P.D. Sharma (Retd.) Prof. Ashu Rani Department of Chemistry Department of Chemistry University of Raj, Jaipur University of Kota, Kota Dr. R.L. Pilaliya, (Retd.) Dr. Sapna Sharma Department of Chemistry, Govt. College Department of Chemistry Bikaner JECRC,university Jaipur Dr. Sanjay Kumar Sharma Dr. Sushil Kumar Sharma Department of Chemistry Department of Chemistry JECRC,university Jaipur University of Kota, Kota Editing and Course Writing Editor Prof. P.S. Verma (Retd.) Dr. Sushil Kumar Sharma Department of Chemistry Assistant Professor, Department of Pure University of Raj, Jaipur and Applied Chemistry University of Kota, Kota Writers: Dr. Shobhana Sharma 1,2,3,6 Dr.Sushil Kr. Sharma 4 ,8,11,14, Assistant Professor, Assistant Professor, Department of Chemistry Department of Pure and S.S.Jain Subodh Applied Chemistry P.G.College,Jaipur University of Kota, Kota Mr. Ankit Sharma 5.7,12 Dr.Girja Shankar Tailor 9,10 Assistant Professor, Assistant Professor, Department of Pure and Department of Chemistry Applied Chemistry Poddar University of Kota, Kota International College, Mansarovar, Jaipur Dr. -
Synthesis of Functionalized Heterocycles Via Palladium Catalyzed Cross-Coupling Reactions
Synthesis of Functionalized Heterocycles via Palladium Catalyzed Cross-Coupling Reactions Dissertation zur Erlangung des akademischen Grades doctor rerum naturalium (Dr. rer. nat.) der Mathematisch-Naturwissenschaftlichen Fakultät der Universität Rostock vorgelegt von Huy Hoang Do, geb. am 31 .07.1987 in Hanoi, Vietnam Rostock, 04.05.2017 Die vorliegende Arbeit wurde in der Zeit von April 2012 bis May 2017 am Institut für Chemie der Universität Rostock am Lehrstuhl für Organische Chemie in der Arbeitsgruppe von Prof. Dr. Peter Langer angefertigt. i 1. Gutachter: Prof. Dr. Peter Langer Universität Rostock, Institut für Chemie 2.Gutachter: Prof. Dr. Jens Christoffers Universität Oldenburg, Institut für Chemie Datum der Einreichung: 03.07.2017 Datum der Verteidigung: 14.11.2017 ERKLÄRUNG Ich versichere hiermit an Eides statt, dass ich die vorliegende Arbeit selbstständig angefertigt und ohne fremde Hilfe verfasst habe. Dazu habe ich keine außer den von mir angegebenen Hilfsmitteln und Quellen verwendet und die den benutzten Werken inhaltlich und wörtlich entnommenen Stellen habe ich als solche kenntlich gemacht. Rostock, 04.05.2017 ………………………. Huy Hoang Do iii Acknowledgements First of all, I would like to gratefully and sincerely thank Prof. Peter Langer (Department of Organic Chemistry, Institute of Chemistry, University of Rostock) who provided me the opportunity to join his research group and for the continuous support of my PhD studies, for his patience and motivation. I am grateful to Dr. Tran Quang Hung who supported me with many advices from the beginning of my work. Beside my supervisor, I would like to thank Dr. Peter Ehlers for sharing his experience and insights which are valuable for my researching process. -

Stille Coupling of Α-Acyloxybenzylstannanes with Acid Chlorides
Stille Coupling of α-Acyloxybenzylstannanes with Acid Chlorides by Junhui Xu A thesis presented to the University of Waterloo in fulfillment of the thesis requirements for the degree of Master of Science in Chemistry Waterloo, Ontario, Canada, 2016 © Junhui Xu 2016 I hereby declare that I am the sole author of this thesis. This is a true copy of the thesis, including any required final revisions, as accepted by my examiners. I understand that my thesis may be made electronically available to the public. ii Abstract Two widely-accepted models (open- and cyclic- transmetalation) are applied to explain the stereochemical outcome of the Stille coupling with organotin compounds containing an sp3 chiral center. However, it is still not possible to predict the stereochemical outcome of this type of Stille coupling before the reaction is conducted. To have a better understanding of the detailed mechanism involved, the Stille couplings of different α-acyloxybenzylstannanes with different acid chlorides were studied in this project. The effects of several factors of both the yield and the stereochemical outcome of the Stille reaction containing an α-acyloxybenzylstannane have been studied. These factors were the protecting group on the organotin nucleophile, ligand, solvent and the substituent on the electrophile. It was found that both a bulky protecting group and ligand with mild σ-donicity could give good yield, while adding any substituent on the electrophile always lowered the yield of reaction. Retention of configuration was observed in the Stille coupling reaction containing an enantiomerically enriched α-alcyloxybenzylstannane. Potential racemization was found after the reaction. Adding a base or doubling the amount of CuCN successfully achieved retention of configuration with > 90% enantiospecificity (e.s.) regardless of the nature of the protecting group, substituent on the electrophile and phosphine ligand. -

A New Class of Organocopper and Organocuprate Compounds Derived from Copper( I) Arenethiolates D
3400 J. Am. Chem. SOC.1992, 114, 3400-3410 A New Class of Organocopper and Organocuprate Compounds Derived from Copper( I) Arenethiolates D. Martin Knotter,t David M. Grove? Wilberth J. J. Smeets,t Anthony L. Spek,t and Gerard van Koten*qt Contribution from the Debye Research Institute, Department of Metal- Mediated Synthesis, University of Utrecht, Padualaan 8, 3584 CH Utrecht, The Netherlands, and Laboratory of Crystallography, University of Utrecht, The Netherlands. Received July 19, 1991 Abstract: Organometallic reagents (ZR; Z = Li, Mg, or Cu; R = Me, CH2SiMe3,or C6H2Me3-2,4,6)react with copper arenethiolates ([CuSArI3;SAr = SC6H3(CH(R’)NMe2)-2-R”-3(1, R’ = R” = H; 2, R’ = Me and R” = H (R-configuration); 3, R’ = H and R” = C1) to give well defined neutral species that have been isolated and characterized both in solution (mol wt determination by cryoscopy and microwave titration, both in benzene, and ‘H NMR) and in the solid state by X-ray crystallography. In these reactions the SAr groups on the copper array are substituted, through interaggregate exchange, by an organic group: [Cu3Li(SAr),R] (13); [Cu,(SAr),R], (4 and 5); [Cu,(SAr)R], (6, 7, and 11); and [Cu4R4][p SAr],[MgSAr], (14). Addition of a phosphine ligand to a solution of ZR and [CuSAr], provides selective formation of [Cu,(SAr),(R)(L)] (8, 9, and 12). The latter are postulated as models for the reactive species in the 1,4-addition reaction of organocuprates with a,p-unsaturated carbonyl compounds (L = alkene function). Crystals of [Cu2(SC6H,(CH2NMe,)- 2}(C6H2Me3-2,4,6)],(6) are monoclinic with a = 13.774 (3) A, b = 13.635 (1) A, c = 19.977 (3) A, 0 = 109.57 (1)O, V = 3535 (1) A3,space group P2,/n, dcalcd= 1.550 g cm-), Z = 4, and R = 0.028 for 6493 reflections with I2 2.5a(I). -
House H O, Respess W L & Whitesides G M. the Chemistry Of
TM CC/NUMBER 2 This Week’s Citation Classic JANUARY 12,1981 House H O, Respess W L & Whitesides G M. The chemistry of carbanions. XII. The role of copper in the conjugate addition of organometallic reagents. J. Org. Chem. 31:312841, 1966. [Dept. Chemistry, Massachusetts Inst. Technol., Cambridge, MA] Preformed methylcopper derivatives have that these copper reagents reacted relatively been found to undergo selective conjugate slowly with nonconjugated ketones and addition to á,âunsaturated carbonyl failed to react at all with nonconjugated compounds. The major reaction path esters and nitriles. Consequently, when these followed in these conjugate additions does organocopper reagents were used in not involve a cyclic sixcentered species stoichiometric quantities, they exhibited high formed from the two reactants. [The SCI® chemoselectivity for conjugate addition even indicates that this paper has been cited in molecules containing other functional over 245 times since 1966.] groups that would have reacted with Grignard reagents. “Other chemists were promptly attracted by Herbert O. House the chemoselectivity offered by Department of Chemistry organocopper reagents and papers Georgia Institute of Technology describing various applications of these Atlanta, GA 30332 reagents in chemical syntheses were published from many laboratories. Within December 2, 1980 five years a sufficient volume of work concerned with organocopper reagents had “Forty years ago small amounts of added been published to justify the preparation of copper compounds were found to favor the an Organic Reactions chapter reviewing the addition of Grignard reagents to the âcarbon subject.3 Publications describing various atom (called conjugate addition) rather than applications of organocopper reagents in the carbonyl group (called 1,2addition, the chemical syntheses have continued to the normal mode of reaction) of á,âunsaturated present. -
Copper-Catalyzed Or Mediated Carbon-Carbon Bond Formation Reactions of Zirconacycles and Alkenylzirconocenes
Copper-Catalyzed or Mediated Carbon-Carbon Bond Formation Reactions of Zirconacycles and Alkenylzirconocenes Martin Kotora, Zhenfeng Xi and Tamotsu Takahashi * Catalysis Research Center and Graduate School of Pharmaceutical Sciences, Hokkaido University, Abstract: In this account we would like to present our recent developments in copper catalyzed or mediated carbon-carbon bond formation reactions of zirconacycles as well as reactions of related alkenylzirconocene compounds. 1. Introduction One of the most attractive reactions of transition metal compounds is coupling (or-dimerization) of alkenes and/or alkynes to give metallacycles. Formally, during this process the metal is oxidized and unsaturated bonds are reduced (Scheme 1). Therefore, in some cases it is called "oxidative coupling", but in other cases it is also called "reductive coupling". This is sometimes confusing. Thus-formed metallacycles such as metallacyclopentanes, metallacyclopentenes and metallacyclopentadienes can participate in a number of further reactions or transformations (ref. 1). Metallacycles prepared from low- valent early transition metal compounds (Zr, Hf, Ti, Nb, Ta etc.) (ref. 2) are very useful since the metal- carbon bonds are easily hydrolyzed by simple acids such as 1N of hydrochloric acid. Organic compounds are readily obtained after hydrolysis. Intramolecular cyclization of diynes, enynes or dienes are known for almost all early transition metals (ref. 1). However, as for selective intermolecular coupling of two different alkynes or alkenes, zirconocene chemistry has a big advantage in this respect (ref. 3). Furthermore, zirconacycles can participate in the catalytic reactions using transmetallation from zirconium to magnesium (ref. 4). Scheme 1 Zirconacyclopentadienes, which can be cleanly prepared from two different alkynes, have long been believed to be very inert for further carbon-carbon bond formation (ref.