Structures and Reactivities of Organocopper Compounds Johann T
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

And C,N-Chelated Organocopper Compounds†
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 2 September 2021 Review C,C- and C,N-Chelated Organocopper Compounds† Liang Liu1, Hui Chen2, Zhenqiang Yang2, Junnian Wei1* and Zhenfeng Xi1,3* 1 Beijing National Laboratory for Molecular Sciences (BNLMS), Key Laboratory of Bioorganic Chemistry and Molecular Engineering of Ministry of Education, College of Chemistry, Peking University, Beijing 100871, China; [email protected] (L.L.), [email protected] (J.W.), [email protected] (Z.X.) 2 Henan Institute of Chemistry Co. Ltd., Henan Academy of Sciences, Zhengzhou 450002, China; [email protected] (H.C.), [email protected] (Z.Y.) 3 State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry (SIOC), Shanghai 200032, China; [email protected] (Z.X.) * Correspondence: [email protected] (J.W.), [email protected] (Z.X.) † Dedicated to Professor Gerard van Koten on the occasion of his 80th birthday Abstract: Copper-catalyzed and organocopper-involved reactions are of great significance in organic synthesis. To have a deep understanding of the reaction mechanisms, the structural characterizations of organocopper intermediates become indispensable. Meanwhile, the structure- function relationship of organocopper compounds would advance rational design and development of new Cu-based reactions and organocopper reagents. Compared to the mono- carbonic ligand, the C,N- and C,C-bidentate ligands better stabilize the unstable organocopper compounds. The bidentate ligands can chelate to the same copper atom via 휂2-mode, forming a mono-cupra-cyclic compounds with at least one acute C-Cu-C angle. When the bidentate ligands bind to two copper atoms via 휂1-mode at each coordinating site, the bimetallic macrocyclic compounds will form nearly linear C-Cu-C angles. -

Conversion of Carbon Dioxide to Acetylene on a Micro Scale
810 NATURE June 14, 1947 Vol. 159 orbitale of the ethmoid is reduced". In the orang Stainless steel was found to be the most satis and the gibbon a large planum orbitale articulates factory furnace material tried. From mild steel in front with the lacrimal, as in man. The figure relatively large amounts of acetylene were produced we give of the orbital wall in Pleaianthropus shows in blank experiments, and a fused silica envelope a condition almost exactly as in man. fitted with a nickel thimble was found, after it had We are here not at present concerned with the been used with calcium and barium metals, to absorb question of whether man and the Australopithecinre carbon dioxide when hot even when no calcium or have arisen from an early anthropoid, or a pre barium was present. In carrying out the absorption anthropoid, or an Old World monkey or a tarsioid ; of carbon dioxide by barium metal in the stainless but we think the evidence afforded by this new skull steel furnace it was found that when the pressure of Plesianthropus shows that the Australopithecinre at which the gas was admitted was less than about and man are very closely allied, and that these small 10·1 mm. of mercury, the yield of acetylene was brained man-like beings were very nearly human. variable and only about 45 per cent. Good yields R. BROOM were obtained when the carbon dioxide at its full J. T. RoBINSON pressure was admitted to the furnace before raising Transvaal Museum, Pretoria. the temperature above 400° C. -

Antifouling Coating Composition, Coating Film Therefrom, Underwater Material Covered with the Coating Film and Antifouling Method
Europäisches Patentamt *EP001342756A1* (19) European Patent Office Office européen des brevets (11) EP 1 342 756 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.7: C09D 5/16 10.09.2003 Bulletin 2003/37 (21) Application number: 03251373.1 (22) Date of filing: 06.03.2003 (84) Designated Contracting States: • Nakamura, Naoya AT BE BG CH CY CZ DE DK EE ES FI FR GB GR Ohtake-shi, Hiroshima (JP) HU IE IT LI LU MC NL PT RO SE SI SK TR • Tsuboi, Makoto Designated Extension States: Ohtake-shi, Hiroshima (JP) AL LT LV MK (74) Representative: Cresswell, Thomas Anthony (30) Priority: 06.03.2002 JP 2002060696 J.A. KEMP & CO. 14 South Square (71) Applicant: CHUGOKU MARINE PAINTS, LTD. Gray’s Inn Ohtake-shi, Hiroshima (JP) London WC1R 5JJ (GB) (72) Inventors: • Oya, Masaaki, Ohtake-shi, Hiroshima (JP) (54) Antifouling coating composition, coating film therefrom, underwater material covered with the coating film and antifouling method (57) An antifouling coating composition comprising: (D) a dehydrating agent. (A) a silyl ester copolymer containing constituent units derived from a polymerizable unsaturated car- boxylic acid silyl ester, (B) a carboxylic acid, (C) a bivalent or trivalent metal compound, and EP 1 342 756 A1 Printed by Jouve, 75001 PARIS (FR) EP 1 342 756 A1 Description FIELD OF THE INVENTION 5 [0001] The present invention relates to an antifouling coating composition which contains a silyl ester copolymer, an antifouling coating film formed from the antifouling coating composition, an antifouling method wherein the antifouling coating composition is used, and a marine vessel (hull) or underwater structure covered with the coating film. -
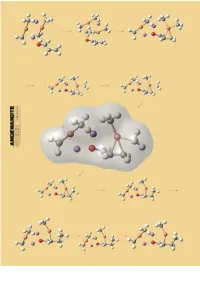
Structures and Reaction Mechanisms of Organocuprate Clusters in Organic Chemistry
REVIEWS Wherefore Art Thou Copper? Structures and Reaction Mechanisms of Organocuprate Clusters in Organic Chemistry Eiichi Nakamura* and Seiji Mori Organocopper reagents provide the principles. This review will summarize example of molecular recognition and most general synthetic tools in organic first the general structural features of supramolecular chemistry, which chemistry for nucleophilic delivery of organocopper compounds and the pre- chemists have long exploited without hard carbanions to electrophilic car- vious mechanistic arguments, and then knowing it. Reasoning about the bon centers. A number of structural describe the most recent mechanistic uniqueness of the copper atom among and mechanistic studies have been pictures obtained through high-level neighboring metal elements in the reported and have led to a wide variety quantum mechanical calculations for periodic table will be presented. of mechanistic proposals, some of three typical organocuprate reactions, which might even be contradictory to carbocupration, conjugate addition, Keywords: catalysis ´ conjugate addi- others. With the recent advent of and SN2 alkylation. The unified view tions ´ copper ´ density functional physical and theoretical methodolo- on the nucleophilic reactivities of met- calculations ´ supramolecular chemis- gies, the accumulated knowledge on al organocuprate clusters thus ob- try organocopper chemistry is being put tained has indicated that organocup- together into a few major mechanistic rate chemistry represents an intricate 1. Introduction 1 R Cu X The desire to learn about the nature of elements has been R or R1 R and will remain a main concern of chemists. In this review, we R Cu will consider what properties of copper make organocopper R1 chemistry so useful in organic chemistry. -

Download Download
— Studies on Lithium Acetylide Kenneth N. Campbell and Barbara K. Campbell, The University of Notre Dame In contrast to the large amount of work done on the acetylene derivatives of sodium, potassium and calcium, little attention has been paid to the analogous compounds of lithium. In 1898 Moissani prepared lithium acetylide on a small scale, by the action of acetylene on a liquid ammonia solution of lithium. He reported that lithium acetylide was less soluble in liquid ammonia than sodium acetylide, and that when isolated from the solvent, it was less stable, undergoing decomposition with evolu- tion of acetylene. On the basis of the weight of lithium acetylide obtained from a given weight of lithium, and from the amount of acetylene liberated on hydrolysis, he assigned to lithium acetylide the formula C2Li2.C2H2.2NH3. Since that time no references to lithium acetylide or lithium alkylacetylides have appeared in the literature. It was the pur- pose of the present work, therefore, to prepare and analyze lithium acetylide and a lithium alkylacetylide, and to compare their reactions with those of the better known sodium derivatives. Experimental Procedure Preparation of Lithium and Sodium Acetylides.—Acetylene gas, washed by bubbling through concentrated sulfuric acid, was passed into two liters of liquid ammonia, while 7 g. (1 mole) of metallic lithium, cut in small pieces, was added gradually, with stirring, at a rate such that the solution did not develop a permanent deep blue color. When the solution became colorless after the addition of the last piece of lithium, the flow of acetylene was stopped. -

Studies Towards a Total Synthesis of Hippeastrine Johanna Myriam Helary 4769139
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by University of East Anglia digital repository Studies towards a total synthesis of Hippeastrine Johanna Myriam Helary 4769139 © I certify that the work contained in the thesis submitted by me for the degree of Master of Science by Research is my original work except where due reference is made to other authors, and has not been previously submitted by me for a degree at this or any other university. 1 Acknowledgements I would like to thanks my mum, my stepdad and Deeptee Horil Roy for their continued supports and encouragements over the last five years. For that I am very grateful. I would also like to thank all of the members Dr G. R Stephenson and Dr C. Richard with special thanks to Dr Julien Doulcet, Dr Paulina Glowacka and the rest of Floor 3 for their help, advices, patient explanations, and good humour over the last two years. Finally I would like to thank Dr. G. R Stephenson for all of his support, guidance and encouragements. In the loving memory of Sarah Delf, devoted friend and colleague. Wherever you are you always will be remembered. Rest in peace my amazing friend and fellow Hippeastrine crew member. 2 Table of Contents Abstract ................................................................................................................................. 5 1. Introduction .......................................................................................................................... 6 1.1. History of alkaloids ....................................................................................................... -

Hoto P QH PH
US010211400B2 (12 ) United States Patent ( 10 ) Patent No. : US 10 ,211 , 400 B2 Hawker et al. ( 45 ) Date of Patent: Feb . 19 , 2019 (54 ) PHOTOPATTERNED GROWTH OF (51 ) Int . Ci. ELECTRONICALLY ACTIVE BRUSH HOIL 51/ 00 ( 2006 . 01 ) POLYMERS FOR LIGHT EMITTING DIODE C09K 11 / 06 (2006 . 01) DISPLAYS (Continued ) (71 ) Applicants : The Regents of the University of ( 52 ) U .S . CI. California , Oakland , CA (US ) ; Rohm CPC . .. .. HOIL 51 /0035 (2013 . 01 ) ; C09K 11/ 06 and Haas Electronic Materials LLC , ( 2013. 01 ) ; HOIL 51/ 0043 ( 2013 .01 ) ; Marlborough , MA (US ) ; Dow Global ( Continued ) Technologies LLC , Midland , MI (US ) (58 ) Field of Classification Search CPC .. .. .. HO1L 51 /0035 ; HO1L 51 /0043 ; HOIL ( 72 ) Inventors : Craig J . Hawker , Santa Barbara , CA (58 ) Fieldof c . 51. .HOZZ /5072 ; HOLLHOIL 551 / 5088 ; HO1L 51/ 5206 ; (US ) ; Zachariah Allen Page , Goleta , CA (US ) ; Peter Trefonas, III, Medway , (Continued ) MA (US ) ; Anatoliy N . Sokolov , Midland , MI (US ) ; John Kramer , ( 56 ) References Cited Midland , MI (US ) ; David S . Laitar, U . S . PATENT DOCUMENTS Midland , MI (US ) ; Sukrit Mukhopadhyay , Midland , MI (US ) ; 6 ,423 , 465 B1 7 /2002 Hawker et al . Benjaporn Narupai, Goleta , CA (US ) ; 6 , 447 , 879 B19 / 2002 Sakurai et al . Christian Wilhelm Pester , Goleta , CA ( Continued ) (US ) OTHER PUBLICATIONS (73 ) Assignees : DOW GLOBAL TECHNOLOGIES LLC , Midland , MI (US ) ; ROHM AND Choi et al. ; “ Simple Detachment Patterning of Organic Layers and HAAS ELECTRONIC MATERIALS Its Application to Organic Light- Emitting Diodes ” ; Adv. Mater. ; 17 , LLC , Marlborough , MA (US ) ; THE No . 2 ; Jan . 31 , 2005 , pp . 166 - 171 . REGENTS OF THE UNIVERSITY (Continued ) OF CALIFORNIA , Oakland, CA (US ) Primary Examiner — Su C Kim ( * ) Notice: Subject to any disclaimer, the term of this ( 74 ) Attorney , Agent , or Firm — Cantor Colburn LLP patent is extended or adjusted under 35 U . -

United States Patent Office Patented Nov
3,111,372 United States Patent Office Patented Nov. 19, 1963 1. 2 earth metal borohydrides, which comprises reacting an 3,111,372 PROCESS FOR THE PRODUCTION OF ALKALI N-trialkyl borazane with an alkali metal acetylide or METAL AND ALKALINE EARTH METAL BORO alkaline earth metal acetylide in the presence of hydrogen HYDRIDES under pressure. For example, N-triethyl borazane can Roland Köster, Mulheim (Ruhr), Germany, assignor to 5 be split up at temperatures higher than 140 C., prefer Studiengesellschaft Kohle m.b.H., Mulheim (Ruhr), ably between 200 and 300° C. by calcium carbide, the Germany acetylide fraction of the calcium carbide becoming free No Drawing. Filed Apr. 28, 1958, Ser. No. 731,125 mostly in the form of ethane. The temperatures used are Claims priority, application Germany Apr. 30, 1957 so low that the initial formation of calcium hydride from 7 Claims. (C. 23-14) O calcium carbide must be considered as not occurring in This invention relates to a process for the production of practice, since the temperatures at which calcium carbide alkali metal and alkaline earth metal borohydrides. reacts with hydrogen alone to form calcium hydride are Belgian patent specification No. 559,053 discloses a substantially higher. Moreover, calcium carbide only process for the production of alkali metal and alkaline reacts easily with hydrogen to form calcium hydride if earth metal borohydrides from borazanes having 3 hydro certain catalysts are present, and in addition the calcium carbon radicals, especially N-trialkyl borazanes, by re hydride formation requires a substantially higher tem action with an alkali metal or alkaline earth metal hy perature than is necessary in the process of the inven dride, with a metal compound of the general formulae tion. -

Mixed Metal Acetylides: the Ptii Aryl Acetylide "[Ptc6h2(Ch2nme2)22,6
FULL PAPER II Mixed Metal Acetylides: The Pt Aryl Acetylide ‘‘[PtC6H2(CH2NMe2)2- 2,6-(C;C)-4]’’ as a Connective Fragment Stephan Back,[a] Robert A. Gossage,[b] Heinrich Lang,*[a] and Gerard van Koten*[b] Keywords: Alkynes / Cyclic voltammetry / Metal-metal interactions / Conjugation / Platinum Using Me3SiC;C{Pt}Cl (1;Me3SiC;C{Pt} = [Pt(C6H2- successful attachment of 1 toaPh3PAu unit leads to linear + {CH2NMe2}2-2,6-{C;CSiMe3}-4] ) a series of platinum Ph3PAuC;C{Pt}Cl (11). Treatment of 11 with FcC;CSnMe3 monoacetylides of the type XC;C{Pt}C;CR [X = SiMe3: 2, produces the heterotrimetallic rigid-rod shaped complex 5 5 R = Ph; 3,R=(η -C5H4)Fe(η -C5H5) (abbreviated as Fc); 4, Ph3PAuC;C{Pt}C;CFc (13). Cyclic voltammetric studies R=C6H4CN-4; 5,R=C6H4(C;CSnMe3)-4;X=H:7,R=Ph; carried out on these Ph3PAu-capped molecules show that the 8,R=Fc;9,R=C6H4CN-4] have been prepared. Studies attachment of an organometallic entity on either side of the II IV directed towards the coordinative properties of the C2 unit of C;C{Pt} fragment leads to a facilitation of the Pt /Pt oxida- 1 have been carried out and heterotrimetallic [µ- tion. (Me3SiC;C{Pt}Cl][Co2(CO)6](10) could be synthesised. The Introduction The growing interest in the application of organometallic Scheme 1. Precursor complex 1 compounds as building blocks for new materials has led to a large number of publications on their synthesis,[1] and the evaluation of theoretical aspects.[2] In particular, the assem- A useful property of 1 is that it possesses two chemically bly, chemistry, and the physical properties of a number of unique reactive sites. -

Decarboxylative Cross-Coupling: Bimetallic Nanoparticles As Catalysts and Low-Temperature Optimisation
Decarboxylative Cross-coupling: Bimetallic Nanoparticles as Catalysts and Low-Temperature Optimisation Joseph Kyle Sheppard Submitted in accordance with the requirements for the degree of Doctor of Philosophy University of Leeds School of Chemistry September 2020 The candidate confirms the work submitted is his own and that appropriate credit has been given where reference has been made to the work of others. I Acknowledgements First and foremost, I would like to thank my supervisors Dr. Bao N. Nguyen and Prof. Patrick McGowan for providing me with the opportunity to undertake this PhD. Bao, you deserve a medal for putting up with me for four whole years! Thank you dearly for everything you have taught me. Patrick, though our meetings were few and far between, I always left them brimming with new ideas and knowledge. I must also thank the wonderful Dr. Alexander Kulak; chiefly for all of your assistance with AAS and SEM analysis but also for being a friendly face that never failed to brighten my days. I’ll deeply miss the hours we spent, shouting over the noise of the spectrometer and trying to work out why it would develop an entirely new issue every time we used it. I would also like to thank Dr. Rebecca Stones for all of her hard work running my TEM samples and teaching me how to analyse the micrographs. Special thanks must also go to Dr. Mary Bayana for her patience in teaching me how to use all of the arcane and intricate analytical machines of the iPRD and to Martin Huscroft for helping me refine my HPLC method. -

Approach and Synthesis of Strychnos Alkaloids
N° d'ordre : 4155 THÈSE Présentée à L'UNIVERSITÉ BORDEAUX I ÉCOLE DOCTORALE DES SCIENCES CHIMIQUES par Dawood Hosni DAWOOD POUR OBTENIR LE GRADE DE DOCTEUR SPÉCIALITÉ : CHIMIE ORGANIQUE ********************* TOWARDS THE SYNTHESIS OF MONOTERPENOIDS INDOLE ALKALOIDS OF THE ASPIDOSPERMATAN AND STRYCHNAN TYPE ********************* Soutenue le: 17 décembre 2010 Après avis de: MM. PIVA Olivier Professeur, Claude Bernard Lyon 1 Rapporteur PALE Patrick Professeur, Louis Pasteur Strasbourg 1 Rapporteur Devant la commission d'examen formée de : MM. PIVA Olivier Professeur, Claude Bernard Lyon 1 Rapporteur PALE Patrick Professeur, Louis Pasteur Strasbourg 1 Rapporteur POISSON Jean-François Chargé de recherche, CNRS Examinateur VINCENT Jean-Marc Directeur de recherche, CNRS Examinateur LANDAIS Yannick Professeur, Bordeaux 1 Directeur de thèse ROBERT Frédéric Chargé de recherche, CNRS Codirecteur de thèse - 2010 - Abbreviations ∆: reflux °C: celsius degrees Ac: acetyle ALB Aluminium Lithium bis(binaphthoxide) complex AIBN : azobis(isobutyronitrile) aq.: aqueous Ar : aromatic BINAP : 2,2'-bis(diphenylphosphino)-1,1'-binaphthyle BINAPO : 2-diphenylphosphino-2'-diphenylphosphinyl-1,1'-binaphthalene BINOL: 1,1’-bi-2-naphthol Boc: tert-butyloxycarbonyle BOX: Bisoxazoline Bz : benzoyle Bn: benzyle cat. : catalytic DBU: 1,8-diazabicyclo[5.4.0]undec-7-ene DCM: dichloromethane DCC: dicyclohexacarbodiimide dr.: diastereomeric ratio DIBAL-H: diisobutylaluminium hydride DIPEA: diisopropyléthylamine (Hünig Base) DMAP: dimethylaminopyridine DME: dimethoxyethane -

Silver-Catalysed Reactions of Alkynes: Recent Advances
Chemical Society Reviews Silver -Catalysed Reactions of Alkynes: Recent Advances Journal: Chemical Society Reviews Manuscript ID: CS-REV-01-2015-000027.R2 Article Type: Review Article Date Submitted by the Author: 02-Jun-2015 Complete List of Authors: Fang, Guichun; Northeast Normal University, Department of Chemistry Bi, Xihe; Northeast Normal University, Page 1 of 48Chem Soc Rev Chemical Society Reviews Dynamic Article Links ► Cite this: DOI: 10.1039/c0xx00000x www.rsc.org/ csr CRITICAL REVIEW Silver-Catalysed Reactions of Alkynes: Recent Advances Guichun Fang,a Xihe Bi*a,b Received (in XXX, XXX) Xth XXXXXXXXX 20XX, Accepted Xth XXXXXXXXX 20XX DOI: 10.1039/b000000x 5 Silver is a less expensive noble metal. Superior alkynophilicity due to π-coordination with the carbon- carbon triple bond, makes silver salts ideal catalysts for alkyne-based organic reactions. This review highlights the progress in alkyne chemistry via silver catalysis primarily over the past five years (ca. 2010–2014). The discussion is developed in terms of the bond type formed with the acetylenic carbon (i.e. , C–C, C–N, C–O, C–Halo, C–P and C–B). Compared with other coinage metals such as Au and Cu, 10 silver catalysis is frequently observed to be unique. This critical review clearly indicates that silver catalysis provides a significant impetus to the rapid evolution of alkyne-based organic reactions, such as alkynylation, hydrofunctionalization, cycloaddition, cycloisomerization, and cascade reactions. alkynylation, cycloaddition, cycloisomerization of functionalized 1. Introduction alkynes (enynes, multiynes, propargyl compounds, etc. ), and hydrofunctionalization.9 Moreover, in addition to the activation Alkynes and their derivatives are among the most valuable 50 of carbon-carbon triple bonds, other functional groups, such as 15 chemical motifs, because of their abundance and versatile 1 imines and carbonyls are also activated through coordination with reactivities.