Rg a Bt\ May 30, 1995
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

And C,N-Chelated Organocopper Compounds†
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 2 September 2021 Review C,C- and C,N-Chelated Organocopper Compounds† Liang Liu1, Hui Chen2, Zhenqiang Yang2, Junnian Wei1* and Zhenfeng Xi1,3* 1 Beijing National Laboratory for Molecular Sciences (BNLMS), Key Laboratory of Bioorganic Chemistry and Molecular Engineering of Ministry of Education, College of Chemistry, Peking University, Beijing 100871, China; [email protected] (L.L.), [email protected] (J.W.), [email protected] (Z.X.) 2 Henan Institute of Chemistry Co. Ltd., Henan Academy of Sciences, Zhengzhou 450002, China; [email protected] (H.C.), [email protected] (Z.Y.) 3 State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry (SIOC), Shanghai 200032, China; [email protected] (Z.X.) * Correspondence: [email protected] (J.W.), [email protected] (Z.X.) † Dedicated to Professor Gerard van Koten on the occasion of his 80th birthday Abstract: Copper-catalyzed and organocopper-involved reactions are of great significance in organic synthesis. To have a deep understanding of the reaction mechanisms, the structural characterizations of organocopper intermediates become indispensable. Meanwhile, the structure- function relationship of organocopper compounds would advance rational design and development of new Cu-based reactions and organocopper reagents. Compared to the mono- carbonic ligand, the C,N- and C,C-bidentate ligands better stabilize the unstable organocopper compounds. The bidentate ligands can chelate to the same copper atom via 휂2-mode, forming a mono-cupra-cyclic compounds with at least one acute C-Cu-C angle. When the bidentate ligands bind to two copper atoms via 휂1-mode at each coordinating site, the bimetallic macrocyclic compounds will form nearly linear C-Cu-C angles. -

The Detection and Determination of Esters
Louisiana State University LSU Digital Commons LSU Historical Dissertations and Theses Graduate School 1958 The etD ection and Determination of Esters. Mohd. Mohsin Qureshi Louisiana State University and Agricultural & Mechanical College Follow this and additional works at: https://digitalcommons.lsu.edu/gradschool_disstheses Recommended Citation Qureshi, Mohd. Mohsin, "The eD tection and Determination of Esters." (1958). LSU Historical Dissertations and Theses. 501. https://digitalcommons.lsu.edu/gradschool_disstheses/501 This Dissertation is brought to you for free and open access by the Graduate School at LSU Digital Commons. It has been accepted for inclusion in LSU Historical Dissertations and Theses by an authorized administrator of LSU Digital Commons. For more information, please contact [email protected]. Copright by Mohcl Mohsin Qureshi 1959 THE DETECTION AND DETERMINATION OF ESTERS A Dissertation Submitted to the Graduate Faculty of the Louisiana State University and Agricultural and Mechanical College in partial fulfillment of the requirements for the degree of Doctor of Philosophy in The Department of Chemistry by Mohd. Mohsin Qureshi M.Sc., Aligarh University, 1944 August, 1958 ACKNOWLEDGMENT The author wishes to express his sincere apprecia tion and gratitude to Dr. Philip W. West under whose guidance this research was carried out. He is grateful to Dr. James G. Traynham for sup plying him with a number of esters and for his many helpful suggestions. The financial support given to him by the Continental Oil Company is gratefully acknowledged. He offers his sincere thanks to Miss Magdalena Usategul for her valuable suggestions and her ungrudging help during the course of this investigation. Dr. Anil K. -

Antifouling Coating Composition, Coating Film Therefrom, Underwater Material Covered with the Coating Film and Antifouling Method
Europäisches Patentamt *EP001342756A1* (19) European Patent Office Office européen des brevets (11) EP 1 342 756 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.7: C09D 5/16 10.09.2003 Bulletin 2003/37 (21) Application number: 03251373.1 (22) Date of filing: 06.03.2003 (84) Designated Contracting States: • Nakamura, Naoya AT BE BG CH CY CZ DE DK EE ES FI FR GB GR Ohtake-shi, Hiroshima (JP) HU IE IT LI LU MC NL PT RO SE SI SK TR • Tsuboi, Makoto Designated Extension States: Ohtake-shi, Hiroshima (JP) AL LT LV MK (74) Representative: Cresswell, Thomas Anthony (30) Priority: 06.03.2002 JP 2002060696 J.A. KEMP & CO. 14 South Square (71) Applicant: CHUGOKU MARINE PAINTS, LTD. Gray’s Inn Ohtake-shi, Hiroshima (JP) London WC1R 5JJ (GB) (72) Inventors: • Oya, Masaaki, Ohtake-shi, Hiroshima (JP) (54) Antifouling coating composition, coating film therefrom, underwater material covered with the coating film and antifouling method (57) An antifouling coating composition comprising: (D) a dehydrating agent. (A) a silyl ester copolymer containing constituent units derived from a polymerizable unsaturated car- boxylic acid silyl ester, (B) a carboxylic acid, (C) a bivalent or trivalent metal compound, and EP 1 342 756 A1 Printed by Jouve, 75001 PARIS (FR) EP 1 342 756 A1 Description FIELD OF THE INVENTION 5 [0001] The present invention relates to an antifouling coating composition which contains a silyl ester copolymer, an antifouling coating film formed from the antifouling coating composition, an antifouling method wherein the antifouling coating composition is used, and a marine vessel (hull) or underwater structure covered with the coating film. -

EI-ICHI NEGISHI Herbert C
MAGICAL POWER OF TRANSITION METALS: PAST, PRESENT, AND FUTURE Nobel Lecture, December 8, 2010 by EI-ICHI NEGISHI Herbert C. Brown Laboratories of Chemistry, Purdue University, 560 Oval Drive, West Lafayette, IN 47907-2084, U.S.A. Not long ago, the primary goal of the synthesis of complex natural products and related compounds of biological and medicinal interest was to be able to synthesize them, preferably before anyone else. While this still remains a very important goal, a number of today’s top-notch synthetic chemists must feel and even think that, given ample resources and time, they are capable of synthesizing virtually all natural products and many analogues thereof. Accepting this notion, what would then be the major goals of organic synthesis in the twenty-first century? One thing appears to be unmistakably certain. Namely, we will always need, perhaps increasingly so with time, the uniquely creative field of synthetic organic and organometallic chemistry to prepare both new and existing organic compounds for the benefit and well-being of mankind. It then seems reasonably clear that, in addition to the question of what compounds to synthesize, that of how best to synthesize them will become increasingly important. As some may have said, the primary goal would then shift from aiming to be the first to synthesize a given compound to seeking its ultimately satisfactory or “last synthesis”. If one carefully goes over various aspects of organic synthetic methodology, one would soon note how primitive and limited it had been until rather recently, or perhaps even today. For the sake of argument, we may propose here that the ultimate goal of organic synthesis is “to be able to synthesize any desired and fundamentally synthesizable organic compounds (a) in high yields, (b) efficiently (in as few steps as possible, for example), (c) selectively, preferably all in t98–99% selectivity, (d) economically, and (e) safely, abbreviated hereafter as the y(es)2 manner.” with or without catalyst R1M + R2X R1R2 + MX R1, R2: carbon groups. -

Recent Progress in the Use of Pd-Catalyzed C-C Cross-Coupling Reactions in the Synthesis of Pharmaceutical Compounds
http://dx.doi.org/10.5935/0103-5053.20140255 J. Braz. Chem. Soc., Vol. 25, No. 12, 2186-2214, 2014. Printed in Brazil - ©2014 Sociedade Brasileira de Química 0103 - 5053 $6.00+0.00 Review Recent Progress in the Use of Pd-Catalyzed C-C Cross-Coupling Reactions in the Synthesis of Pharmaceutical Compounds André F. P. Biajoli, Cristiane S. Schwalm, Jones Limberger, Thiago S. Claudino and Adriano L. Monteiro* Laboratory of Molecular Catalysis, Institute of Chemistry, UFRGS, Av. Bento Gonçalves, 9500, 91501-970 Porto Alegre-RS, Brazil A grande capacidade do paládio de formar ligações carbono-carbono entre substratos apropriadamente funcionalizados permitiu que os químicos orgânicos efetuassem transformações antes impossíveis ou alcançáveis somente através de rotas muito longas. Neste contexto, uma das mais elegantes e importantes aplicações das reações de acoplamento cruzado catalisadas por paládio é a síntese de compostos de interesse farmacêutico. A presente revisão tem por objetivo apresentar uma visão geral do uso de acoplamentos cruzados na síntese de componentes de medicamentos (ou de candidatos a medicamentos), independentemente da escala, compreendendo o período de 2011 até o final de julho de 2014. The impressive ability of palladium to assemble C-C bonds between appropriately functionalized substrates has allowed synthetic organic chemists to perform transformations that were previously impossible or only possible using multi-step approaches. In this context, one of the most important and elegant applications of the Pd-catalyzed C-C coupling reactions currently is the synthesis of pharmaceuticals. This review is intended to give a picture of the applications of Pd-catalyzed C-C cross-coupling reactions for the synthesis of drug components or drug candidates regardless of the scale from 2011 through to the end of July, 2014. -
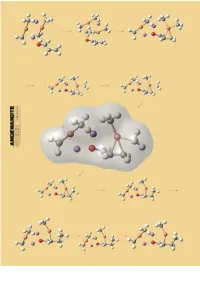
Structures and Reaction Mechanisms of Organocuprate Clusters in Organic Chemistry
REVIEWS Wherefore Art Thou Copper? Structures and Reaction Mechanisms of Organocuprate Clusters in Organic Chemistry Eiichi Nakamura* and Seiji Mori Organocopper reagents provide the principles. This review will summarize example of molecular recognition and most general synthetic tools in organic first the general structural features of supramolecular chemistry, which chemistry for nucleophilic delivery of organocopper compounds and the pre- chemists have long exploited without hard carbanions to electrophilic car- vious mechanistic arguments, and then knowing it. Reasoning about the bon centers. A number of structural describe the most recent mechanistic uniqueness of the copper atom among and mechanistic studies have been pictures obtained through high-level neighboring metal elements in the reported and have led to a wide variety quantum mechanical calculations for periodic table will be presented. of mechanistic proposals, some of three typical organocuprate reactions, which might even be contradictory to carbocupration, conjugate addition, Keywords: catalysis ´ conjugate addi- others. With the recent advent of and SN2 alkylation. The unified view tions ´ copper ´ density functional physical and theoretical methodolo- on the nucleophilic reactivities of met- calculations ´ supramolecular chemis- gies, the accumulated knowledge on al organocuprate clusters thus ob- try organocopper chemistry is being put tained has indicated that organocup- together into a few major mechanistic rate chemistry represents an intricate 1. Introduction 1 R Cu X The desire to learn about the nature of elements has been R or R1 R and will remain a main concern of chemists. In this review, we R Cu will consider what properties of copper make organocopper R1 chemistry so useful in organic chemistry. -

The 2010 Chemistry Nobel Prize: Pd(0)-Catalyzed Organic Synthesis
GENERAL ARTICLE The 2010 Chemistry Nobel Prize: Pd(0)-Catalyzed Organic Synthesis Gopalpur Nagendrappa and Y C Sunil Kumar The 2010 Nobel Prize in Chemistry was awarded to three scientists, R F Heck, E-I Negishi and A Suzuki, for their work on “Palladium – Catalyzed Cross Couplings in Organic Syn- (left) G Nagendrappa thesis”. It pertains to research done over a period of four was a Professor of decades. The synthetic procedures embodied in their work Organic Chemistry at enable construction of C–C bond selectively between complex Bangalore University, molecules as in simple ones at desired positions without and Head of the Department of Medici- disturbing any functional groups at other parts of the reacting nal Chemistry, Sri molecules. The work finds wide applications in the synthesis Ramachandra (Medical) of pharmaceuticals, agricultural chemicals, and molecules for University, Chennai. electronics and other applications. It would not have been He is currently in Jain University, Bangalore. possible to synthesize some of the complex natural products or He continues to teach synthetic compounds without using these coupling reactions and do research. His in one or more steps. work is in the area of organosilicon chemis- Introduction try, synthetic and mechanistic organic In mythical stories and folk tales we come across characters that, chemistry, and clay- catalysed organic while uttering some manthras (words of charm), throw a pinch or reactions (Green fistful of a magic powder, and suddenly there appears the object Chemistry). or person they wished for or an event happens the way they want. (right) Sunil Kumar is A large number of movies have been made with such themes and a PhD from Mysore characters that would be depicted as ‘scientists’. -

MICROREVIEW Reactivity of Polar Organometallic Compounds In
MICROREVIEW Reactivity of Polar Organometallic Compounds in Unconventional Reaction Media: Challenges and Opportunities Joaquin García-Álvarez,*[a] Eva Hevia,*[b] and Vito Capriati*[c] This paper is gratefully dedicated to the memory of Dr. Guy Lavigne Abstract: Developing new green solvents in designing chemical chemicals in chemical transformations is, indeed, solvents used products and processes or successfully employing the already as reaction media, which account for 80–90% of mass utilization existing ones is one of the key subjects in Green Chemistry and is in a typical pharmaceutical/fine chemical operational process. especially important in Organometallic Chemistry, which is an Thus, the solvent itself is often a critical parameter especially in interdisciplinary field. Can we advantageously use unconventional drug product manufacturing and is as well responsible for most reaction media in place of current harsh organic solvents also for waste generated in the chemical industries and laboratories.[3] polar organometallic compounds? This Microreview critically Following these considerations, some of the most critical analyses the state-of-the-art on this topic and showcases recent and intriguing questions that arise are: Can we get traditional developments and breakthroughs which are becoming new research organic solvents out of organometallic reactions?[4] Can we use directions in this field. Because metals cover a vast swath of the protic, recyclable, biodegradable, and cheap unconventional periodic table, the content is organised into three Sections solvents also for highly reactive organometallic compounds? discussing the reactivity of organometallic compounds of s-, p- and Answering these questions would not only mean to break new d-block elements in unconventional solvents. -

New Preparations and Reactions of Salt Stabilized Organozinc Reagents For
Dissertation zur Erlangung des Doktorgrades der Fakultät für Chemie und Pharmazie der Ludwig-Maximilians-Universität München NEW PREPARATIONS AND REACTIONS OF SALT STABILIZED ORGANOZINC REAGENTS FOR THE FUNCTIONALIZATION OF AROMATICS, HETEROAROMATICS, AND ALLYLIC COMPOUNDS von Mario Ferdinand Ellwart aus München 2016 ERKLÄRUNG Diese Dissertation wurde im Sinne von § 7 der Promotionsordnung vom 28. November 2011 von Herrn Prof. Dr. Paul Knochel betreut. EIDESSTATTLICHE VERSICHERUNG Diese Dissertation wurde eigenständig und ohne unerlaubte Hilfe erarbeitet. München, ………………………. ………….……………………………… (Mario F. Ellwart) Dissertation eingereicht am: 07.10.2016 1. Gutachter: Prof. Dr. Paul Knochel 2. Gutachter: Prof. Dr. Oliver Trapp Mündliche Prüfung am: 06.12.2016 This work was carried out from January 2014 to October 2016 under the guidance of Prof. Dr. Paul Knochel at the Department of Chemistry of the Ludwig-Maximilians-Universität, Munich. Firstly, I would like to express my appreciation to Prof. Dr. Paul Knochel for giving me the great opportunity to do my Ph.D. in his group and for his guidance in the course of my scientific research. I am also very grateful to Prof. Dr. Oliver Trapp for agreeing to be the second reviewer of this thesis, as well as Prof. Dr. Manfred Heuschmann, Prof. Dr. Klaus T. Wanner, Prof. Dr. Konstantin Karaghiosoff, and Prof. Dr. Regina de Vivie-Riedle for their interest shown in this manuscript by accepting to be referees. I also would like to thank Michael Eisold, Marthe Ketels, Meike Simon, and Dr. Benedikt S. Soller for the careful correction of this manuscript. I thank all past and present co-workers I have met in the Knochel group for their kindness and their help. -

Pacs by Chemical Name (Mg/M3) (Pdf)
Table 4: Protective Action Criteria (PAC) Rev 25 based on applicable 60-minute AEGLs, ERPGs, or TEELs. Values are presented in mg/m3. August 2009 Table 4 is an alphabetical listing of the chemicals in the PAC data set. It provides Chemical Abstract Service Registry Numbers (CASRNs)1, PAC values, and technical information on the source of the PAC values. Table 4 presents all values for TEEL-0, PAC-1, PAC-2, and PAC-3 in mg/m3. The conversion of ppm to mg/m3 is calculated assuming 25 ºC and 760 mm Hg. The columns presented in Table 4 provide the following information: Heading Definition No. The ordered numbering of the chemicals as they appear in this alphabetical listing. Chemical Name The chemical name given to the PAC Development Team. CASRN The Chemical Abstract Service Registry Number for this chemical. TEEL-0 This is the threshold concentration below which most people will experience no adverse health effects. This PAC is always based on TEEL-0 because AEGL-0 or ERPG-0 values do not exist. PAC-1 Based on the applicable AEGL-1, ERPG-1, or TEEL-1 value. PAC-2 Based on the applicable AEGL-2, ERPG-2, or TEEL-2 value. PAC-3 Based on the applicable AEGL-3, ERPG-3, or TEEL-3 value. Source of PACs: Technical comments provided by the PAC development team that TEEL-0, PAC-1, indicate the source of the data used to derive PAC values. Future efforts PAC-2, PAC-3 are directed at reviewing, revising, and enhancing this information. -

Studies Towards a Total Synthesis of Hippeastrine Johanna Myriam Helary 4769139
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by University of East Anglia digital repository Studies towards a total synthesis of Hippeastrine Johanna Myriam Helary 4769139 © I certify that the work contained in the thesis submitted by me for the degree of Master of Science by Research is my original work except where due reference is made to other authors, and has not been previously submitted by me for a degree at this or any other university. 1 Acknowledgements I would like to thanks my mum, my stepdad and Deeptee Horil Roy for their continued supports and encouragements over the last five years. For that I am very grateful. I would also like to thank all of the members Dr G. R Stephenson and Dr C. Richard with special thanks to Dr Julien Doulcet, Dr Paulina Glowacka and the rest of Floor 3 for their help, advices, patient explanations, and good humour over the last two years. Finally I would like to thank Dr. G. R Stephenson for all of his support, guidance and encouragements. In the loving memory of Sarah Delf, devoted friend and colleague. Wherever you are you always will be remembered. Rest in peace my amazing friend and fellow Hippeastrine crew member. 2 Table of Contents Abstract ................................................................................................................................. 5 1. Introduction .......................................................................................................................... 6 1.1. History of alkaloids ....................................................................................................... -

Hoto P QH PH
US010211400B2 (12 ) United States Patent ( 10 ) Patent No. : US 10 ,211 , 400 B2 Hawker et al. ( 45 ) Date of Patent: Feb . 19 , 2019 (54 ) PHOTOPATTERNED GROWTH OF (51 ) Int . Ci. ELECTRONICALLY ACTIVE BRUSH HOIL 51/ 00 ( 2006 . 01 ) POLYMERS FOR LIGHT EMITTING DIODE C09K 11 / 06 (2006 . 01) DISPLAYS (Continued ) (71 ) Applicants : The Regents of the University of ( 52 ) U .S . CI. California , Oakland , CA (US ) ; Rohm CPC . .. .. HOIL 51 /0035 (2013 . 01 ) ; C09K 11/ 06 and Haas Electronic Materials LLC , ( 2013. 01 ) ; HOIL 51/ 0043 ( 2013 .01 ) ; Marlborough , MA (US ) ; Dow Global ( Continued ) Technologies LLC , Midland , MI (US ) (58 ) Field of Classification Search CPC .. .. .. HO1L 51 /0035 ; HO1L 51 /0043 ; HOIL ( 72 ) Inventors : Craig J . Hawker , Santa Barbara , CA (58 ) Fieldof c . 51. .HOZZ /5072 ; HOLLHOIL 551 / 5088 ; HO1L 51/ 5206 ; (US ) ; Zachariah Allen Page , Goleta , CA (US ) ; Peter Trefonas, III, Medway , (Continued ) MA (US ) ; Anatoliy N . Sokolov , Midland , MI (US ) ; John Kramer , ( 56 ) References Cited Midland , MI (US ) ; David S . Laitar, U . S . PATENT DOCUMENTS Midland , MI (US ) ; Sukrit Mukhopadhyay , Midland , MI (US ) ; 6 ,423 , 465 B1 7 /2002 Hawker et al . Benjaporn Narupai, Goleta , CA (US ) ; 6 , 447 , 879 B19 / 2002 Sakurai et al . Christian Wilhelm Pester , Goleta , CA ( Continued ) (US ) OTHER PUBLICATIONS (73 ) Assignees : DOW GLOBAL TECHNOLOGIES LLC , Midland , MI (US ) ; ROHM AND Choi et al. ; “ Simple Detachment Patterning of Organic Layers and HAAS ELECTRONIC MATERIALS Its Application to Organic Light- Emitting Diodes ” ; Adv. Mater. ; 17 , LLC , Marlborough , MA (US ) ; THE No . 2 ; Jan . 31 , 2005 , pp . 166 - 171 . REGENTS OF THE UNIVERSITY (Continued ) OF CALIFORNIA , Oakland, CA (US ) Primary Examiner — Su C Kim ( * ) Notice: Subject to any disclaimer, the term of this ( 74 ) Attorney , Agent , or Firm — Cantor Colburn LLP patent is extended or adjusted under 35 U .