Srinivasan College of Arts and Science Perambalur
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

And C,N-Chelated Organocopper Compounds†
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 2 September 2021 Review C,C- and C,N-Chelated Organocopper Compounds† Liang Liu1, Hui Chen2, Zhenqiang Yang2, Junnian Wei1* and Zhenfeng Xi1,3* 1 Beijing National Laboratory for Molecular Sciences (BNLMS), Key Laboratory of Bioorganic Chemistry and Molecular Engineering of Ministry of Education, College of Chemistry, Peking University, Beijing 100871, China; [email protected] (L.L.), [email protected] (J.W.), [email protected] (Z.X.) 2 Henan Institute of Chemistry Co. Ltd., Henan Academy of Sciences, Zhengzhou 450002, China; [email protected] (H.C.), [email protected] (Z.Y.) 3 State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry (SIOC), Shanghai 200032, China; [email protected] (Z.X.) * Correspondence: [email protected] (J.W.), [email protected] (Z.X.) † Dedicated to Professor Gerard van Koten on the occasion of his 80th birthday Abstract: Copper-catalyzed and organocopper-involved reactions are of great significance in organic synthesis. To have a deep understanding of the reaction mechanisms, the structural characterizations of organocopper intermediates become indispensable. Meanwhile, the structure- function relationship of organocopper compounds would advance rational design and development of new Cu-based reactions and organocopper reagents. Compared to the mono- carbonic ligand, the C,N- and C,C-bidentate ligands better stabilize the unstable organocopper compounds. The bidentate ligands can chelate to the same copper atom via 휂2-mode, forming a mono-cupra-cyclic compounds with at least one acute C-Cu-C angle. When the bidentate ligands bind to two copper atoms via 휂1-mode at each coordinating site, the bimetallic macrocyclic compounds will form nearly linear C-Cu-C angles. -

NBO Applications, 2020
NBO Bibliography 2020 2531 publications – Revised and compiled by Ariel Andrea on Aug. 9, 2021 Aarabi, M.; Gholami, S.; Grabowski, S. J. S-H ... O and O-H ... O Hydrogen Bonds-Comparison of Dimers of Thiocarboxylic and Carboxylic Acids Chemphyschem, (21): 1653-1664 2020. 10.1002/cphc.202000131 Aarthi, K. V.; Rajagopal, H.; Muthu, S.; Jayanthi, V.; Girija, R. Quantum chemical calculations, spectroscopic investigation and molecular docking analysis of 4-chloro- N-methylpyridine-2-carboxamide Journal of Molecular Structure, (1210) 2020. 10.1016/j.molstruc.2020.128053 Abad, N.; Lgaz, H.; Atioglu, Z.; Akkurt, M.; Mague, J. T.; Ali, I. H.; Chung, I. M.; Salghi, R.; Essassi, E.; Ramli, Y. Synthesis, crystal structure, hirshfeld surface analysis, DFT computations and molecular dynamics study of 2-(benzyloxy)-3-phenylquinoxaline Journal of Molecular Structure, (1221) 2020. 10.1016/j.molstruc.2020.128727 Abbenseth, J.; Wtjen, F.; Finger, M.; Schneider, S. The Metaphosphite (PO2-) Anion as a Ligand Angewandte Chemie-International Edition, (59): 23574-23578 2020. 10.1002/anie.202011750 Abbenseth, J.; Goicoechea, J. M. Recent developments in the chemistry of non-trigonal pnictogen pincer compounds: from bonding to catalysis Chemical Science, (11): 9728-9740 2020. 10.1039/d0sc03819a Abbenseth, J.; Schneider, S. A Terminal Chlorophosphinidene Complex Zeitschrift Fur Anorganische Und Allgemeine Chemie, (646): 565-569 2020. 10.1002/zaac.202000010 Abbiche, K.; Acharjee, N.; Salah, M.; Hilali, M.; Laknifli, A.; Komiha, N.; Marakchi, K. Unveiling the mechanism and selectivity of 3+2 cycloaddition reactions of benzonitrile oxide to ethyl trans-cinnamate, ethyl crotonate and trans-2-penten-1-ol through DFT analysis Journal of Molecular Modeling, (26) 2020. -

Contemporary Organosilicon Chemistry
Contemporary organosilicon chemistry Edited by Steve Marsden Generated on 05 October 2021, 02:13 Imprint Beilstein Journal of Organic Chemistry www.bjoc.org ISSN 1860-5397 Email: [email protected] The Beilstein Journal of Organic Chemistry is published by the Beilstein-Institut zur Förderung der Chemischen Wissenschaften. This thematic issue, published in the Beilstein Beilstein-Institut zur Förderung der Journal of Organic Chemistry, is copyright the Chemischen Wissenschaften Beilstein-Institut zur Förderung der Chemischen Trakehner Straße 7–9 Wissenschaften. The copyright of the individual 60487 Frankfurt am Main articles in this document is the property of their Germany respective authors, subject to a Creative www.beilstein-institut.de Commons Attribution (CC-BY) license. Contemporary organosilicon chemistry Steve Marsden Editorial Open Access Address: Beilstein Journal of Organic Chemistry 2007, 3, No. 4. School of Chemistry, University of Leeds, Leeds LS2 9JT, UK doi:10.1186/1860-5397-3-4 Email: Received: 06 February 2007 Steve Marsden - [email protected] Accepted: 08 February 2007 Published: 08 February 2007 © 2007 Marsden; licensee Beilstein-Institut License and terms: see end of document. Abstract Editorial for the Thematic Series on Contemporary Organosilicon Chemistry. The field of organosilicon chemistry has a rich and varied the 1990s, and equivalent to the number appearing in the much history, and has long since made the progression from chemical longer established field of organoboron chemistry -

Herbert Charles Brown, a Biographical Memoir
NATIONAL ACADEMY OF SCIENCES H E R B E R T Ch ARLES BROWN 1 9 1 2 — 2 0 0 4 A Biographical Memoir by E I-I CH I N EGIS HI Any opinions expressed in this memoir are those of the author and do not necessarily reflect the views of the National Academy of Sciences. Biographical Memoir COPYRIGHT 2008 NATIONAL ACADEMY OF SCIENCES WASHINGTON, D.C. Photograph Credit Here. HERBERT CHARLES BROWN May 22, 1912–December 19, 2004 BY EI -ICH I NEGISHI ERBERT CHARLES BROWN, R. B. Wetherill Research Profes- Hsor Emeritus of Purdue University and one of the truly pioneering giants in the field of organic-organometallic chemistry, died of a heart attack on December 19, 2004, at age 92. As it so happened, this author visited him at his home to discuss with him an urgent chemistry-related matter only about 10 hours before his death. For his age he appeared well, showing no sign of his sudden death the next morn- ing. His wife, Sarah Baylen Brown, 89, followed him on May 29, 2005. They were survived by their only child, Charles A. Brown of Hitachi Ltd. and his family. H. C. Brown shared the Nobel Prize in Chemistry in 1979 with G. Wittig of Heidelberg, Germany. Their pioneering explorations of boron chemistry and phosphorus chemistry, respectively, were recognized. Aside from several biochemists, including V. du Vigneaud in 1955, H. C. Brown was only the second American organic chemist to win a Nobel Prize behind R. B. Woodward, in 1965. His several most significant contribu- tions in the area of boron chemistry include (1) codiscovery of sodium borohyride (1972[1], pp. -

Antifouling Coating Composition, Coating Film Therefrom, Underwater Material Covered with the Coating Film and Antifouling Method
Europäisches Patentamt *EP001342756A1* (19) European Patent Office Office européen des brevets (11) EP 1 342 756 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.7: C09D 5/16 10.09.2003 Bulletin 2003/37 (21) Application number: 03251373.1 (22) Date of filing: 06.03.2003 (84) Designated Contracting States: • Nakamura, Naoya AT BE BG CH CY CZ DE DK EE ES FI FR GB GR Ohtake-shi, Hiroshima (JP) HU IE IT LI LU MC NL PT RO SE SI SK TR • Tsuboi, Makoto Designated Extension States: Ohtake-shi, Hiroshima (JP) AL LT LV MK (74) Representative: Cresswell, Thomas Anthony (30) Priority: 06.03.2002 JP 2002060696 J.A. KEMP & CO. 14 South Square (71) Applicant: CHUGOKU MARINE PAINTS, LTD. Gray’s Inn Ohtake-shi, Hiroshima (JP) London WC1R 5JJ (GB) (72) Inventors: • Oya, Masaaki, Ohtake-shi, Hiroshima (JP) (54) Antifouling coating composition, coating film therefrom, underwater material covered with the coating film and antifouling method (57) An antifouling coating composition comprising: (D) a dehydrating agent. (A) a silyl ester copolymer containing constituent units derived from a polymerizable unsaturated car- boxylic acid silyl ester, (B) a carboxylic acid, (C) a bivalent or trivalent metal compound, and EP 1 342 756 A1 Printed by Jouve, 75001 PARIS (FR) EP 1 342 756 A1 Description FIELD OF THE INVENTION 5 [0001] The present invention relates to an antifouling coating composition which contains a silyl ester copolymer, an antifouling coating film formed from the antifouling coating composition, an antifouling method wherein the antifouling coating composition is used, and a marine vessel (hull) or underwater structure covered with the coating film. -

EI-ICHI NEGISHI Herbert C
MAGICAL POWER OF TRANSITION METALS: PAST, PRESENT, AND FUTURE Nobel Lecture, December 8, 2010 by EI-ICHI NEGISHI Herbert C. Brown Laboratories of Chemistry, Purdue University, 560 Oval Drive, West Lafayette, IN 47907-2084, U.S.A. Not long ago, the primary goal of the synthesis of complex natural products and related compounds of biological and medicinal interest was to be able to synthesize them, preferably before anyone else. While this still remains a very important goal, a number of today’s top-notch synthetic chemists must feel and even think that, given ample resources and time, they are capable of synthesizing virtually all natural products and many analogues thereof. Accepting this notion, what would then be the major goals of organic synthesis in the twenty-first century? One thing appears to be unmistakably certain. Namely, we will always need, perhaps increasingly so with time, the uniquely creative field of synthetic organic and organometallic chemistry to prepare both new and existing organic compounds for the benefit and well-being of mankind. It then seems reasonably clear that, in addition to the question of what compounds to synthesize, that of how best to synthesize them will become increasingly important. As some may have said, the primary goal would then shift from aiming to be the first to synthesize a given compound to seeking its ultimately satisfactory or “last synthesis”. If one carefully goes over various aspects of organic synthetic methodology, one would soon note how primitive and limited it had been until rather recently, or perhaps even today. For the sake of argument, we may propose here that the ultimate goal of organic synthesis is “to be able to synthesize any desired and fundamentally synthesizable organic compounds (a) in high yields, (b) efficiently (in as few steps as possible, for example), (c) selectively, preferably all in t98–99% selectivity, (d) economically, and (e) safely, abbreviated hereafter as the y(es)2 manner.” with or without catalyst R1M + R2X R1R2 + MX R1, R2: carbon groups. -

FAROOK COLLEGE (Autonomous)
FAROOK COLLEGE (Autonomous) M.Sc. DEGREE PROGRAMME IN CHEMISTRY CHOICE BASED CREDIT AND SEMESTER SYSTEM-PG (FCCBCSSPG-2019) SCHEME AND SYLLABI 2019 ADMISSION ONWARDS 1 CERTIFICATE I hereby certify that the documents attached are the bona fide copies of the syllabus of M.Sc. Chemistry Programme to be effective from the academic year 2019-20 onwards. Date: Place: P R I N C I P A L 2 FAROOK COLLEGE (AUTONOMOUS) MSc. CHEMISTRY (CSS PATTERN) Regulations and Syllabus with effect from 2019 admission Pattern of the Programme a. The name of the programme shall be M.Sc. Chemistry under CSS pattern. b. The programme shall be offered in four semesters within a period of two academic years. c. Eligibility for admission will be as per the rules laid down by the College from time to time. d. Details of the courses offered for the programme are given in Table 1. The programme shall be conducted in accordance with the programme pattern, scheme of examination and syllabus prescribed. Of the 25 hours per week, 13 hours shall be allotted for theory and 12 hours for practical; 1 theory hour per week during even semesters shall be allotted for seminar. Theory Courses In the first three semesters, there will be four theory courses; and in the fourth semester, three theory courses. All the theory courses in the first and second semesters are core courses. In the third semester there will be three core theory courses and one elective theory course. College can choose any one of the elective courses given in Table 1. -

Organometallic Compounds
Chapter 11 Organometallic compounds Organometallics Reactions using organometallics Organometallic compounds Ch 11 #2 = comp’s containing a carbon-metal bond δ− δ+ C in organometallic comp’ds are nucleophilic. C in organic comp’d (like ROH, RNH , and RX) 2 δ+ δ are electrophilic. − due to ∆EN Ch 11 #3 in substitution reactions a carbon Nu: R-Li and R-MgX Ch 11 #4 (used to be) the two most common organometallics organolithium comp’ds BuLi = an alkyl lithium organomagnesium comp’ds = Grignard reagents 1912 Nobel Prize R, Ar, vinyl all possible; Br (as X) popular Ether (solvent) coordinates Mg, stabilizing it. Reactions of R-Li and R-MgX Ch 11 #5 reacts like a carbanion C:– ~ a C Nu: reactions as C Nu: (like SN(2)) nucleophilic addition to carbonyls ~ more often Chapt 16 Ch 11 #6 R-Li and R-MgX are very strong B: how strong? pKa? − − react even with very weak acid stronger than OH? NH2? useful for preparing deuterated HC Storage and reaction must be acid- and moisture-free. Transmetal(l)ation Ch 11 #7 R-Li is more reactive than R-MgX is. C–Li more polar than C–Mg C of R-Li more nu-philic [better Nu:] transmetalation [metal exchange] to less reactive [more stable] organometallic Coupling using Gilman reagent Ch 11 #8 coupling reaction (in organic chemistry) two hydrocarbon fragments are coupled (to form C–C) with the aid of a (transition) metal catalyst Gilman reagent coupling of R of R-X and R’ of Gilman reagent RX + R’2CuLi R–R’ mechanism? substitution of X with R’? not clear Ch 11 #9 R can be alkyl, aryl, or alkenyl [vinyl] which is not possible by R-Li or R-MgX Is R of Gilman why? they are SN(2). -

Recent Progress in the Use of Pd-Catalyzed C-C Cross-Coupling Reactions in the Synthesis of Pharmaceutical Compounds
http://dx.doi.org/10.5935/0103-5053.20140255 J. Braz. Chem. Soc., Vol. 25, No. 12, 2186-2214, 2014. Printed in Brazil - ©2014 Sociedade Brasileira de Química 0103 - 5053 $6.00+0.00 Review Recent Progress in the Use of Pd-Catalyzed C-C Cross-Coupling Reactions in the Synthesis of Pharmaceutical Compounds André F. P. Biajoli, Cristiane S. Schwalm, Jones Limberger, Thiago S. Claudino and Adriano L. Monteiro* Laboratory of Molecular Catalysis, Institute of Chemistry, UFRGS, Av. Bento Gonçalves, 9500, 91501-970 Porto Alegre-RS, Brazil A grande capacidade do paládio de formar ligações carbono-carbono entre substratos apropriadamente funcionalizados permitiu que os químicos orgânicos efetuassem transformações antes impossíveis ou alcançáveis somente através de rotas muito longas. Neste contexto, uma das mais elegantes e importantes aplicações das reações de acoplamento cruzado catalisadas por paládio é a síntese de compostos de interesse farmacêutico. A presente revisão tem por objetivo apresentar uma visão geral do uso de acoplamentos cruzados na síntese de componentes de medicamentos (ou de candidatos a medicamentos), independentemente da escala, compreendendo o período de 2011 até o final de julho de 2014. The impressive ability of palladium to assemble C-C bonds between appropriately functionalized substrates has allowed synthetic organic chemists to perform transformations that were previously impossible or only possible using multi-step approaches. In this context, one of the most important and elegant applications of the Pd-catalyzed C-C coupling reactions currently is the synthesis of pharmaceuticals. This review is intended to give a picture of the applications of Pd-catalyzed C-C cross-coupling reactions for the synthesis of drug components or drug candidates regardless of the scale from 2011 through to the end of July, 2014. -
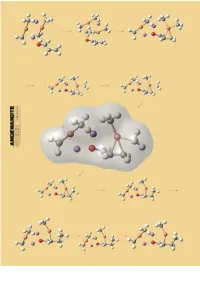
Structures and Reaction Mechanisms of Organocuprate Clusters in Organic Chemistry
REVIEWS Wherefore Art Thou Copper? Structures and Reaction Mechanisms of Organocuprate Clusters in Organic Chemistry Eiichi Nakamura* and Seiji Mori Organocopper reagents provide the principles. This review will summarize example of molecular recognition and most general synthetic tools in organic first the general structural features of supramolecular chemistry, which chemistry for nucleophilic delivery of organocopper compounds and the pre- chemists have long exploited without hard carbanions to electrophilic car- vious mechanistic arguments, and then knowing it. Reasoning about the bon centers. A number of structural describe the most recent mechanistic uniqueness of the copper atom among and mechanistic studies have been pictures obtained through high-level neighboring metal elements in the reported and have led to a wide variety quantum mechanical calculations for periodic table will be presented. of mechanistic proposals, some of three typical organocuprate reactions, which might even be contradictory to carbocupration, conjugate addition, Keywords: catalysis ´ conjugate addi- others. With the recent advent of and SN2 alkylation. The unified view tions ´ copper ´ density functional physical and theoretical methodolo- on the nucleophilic reactivities of met- calculations ´ supramolecular chemis- gies, the accumulated knowledge on al organocuprate clusters thus ob- try organocopper chemistry is being put tained has indicated that organocup- together into a few major mechanistic rate chemistry represents an intricate 1. Introduction 1 R Cu X The desire to learn about the nature of elements has been R or R1 R and will remain a main concern of chemists. In this review, we R Cu will consider what properties of copper make organocopper R1 chemistry so useful in organic chemistry. -

The 2010 Chemistry Nobel Prize: Pd(0)-Catalyzed Organic Synthesis
GENERAL ARTICLE The 2010 Chemistry Nobel Prize: Pd(0)-Catalyzed Organic Synthesis Gopalpur Nagendrappa and Y C Sunil Kumar The 2010 Nobel Prize in Chemistry was awarded to three scientists, R F Heck, E-I Negishi and A Suzuki, for their work on “Palladium – Catalyzed Cross Couplings in Organic Syn- (left) G Nagendrappa thesis”. It pertains to research done over a period of four was a Professor of decades. The synthetic procedures embodied in their work Organic Chemistry at enable construction of C–C bond selectively between complex Bangalore University, molecules as in simple ones at desired positions without and Head of the Department of Medici- disturbing any functional groups at other parts of the reacting nal Chemistry, Sri molecules. The work finds wide applications in the synthesis Ramachandra (Medical) of pharmaceuticals, agricultural chemicals, and molecules for University, Chennai. electronics and other applications. It would not have been He is currently in Jain University, Bangalore. possible to synthesize some of the complex natural products or He continues to teach synthetic compounds without using these coupling reactions and do research. His in one or more steps. work is in the area of organosilicon chemis- Introduction try, synthetic and mechanistic organic In mythical stories and folk tales we come across characters that, chemistry, and clay- catalysed organic while uttering some manthras (words of charm), throw a pinch or reactions (Green fistful of a magic powder, and suddenly there appears the object Chemistry). or person they wished for or an event happens the way they want. (right) Sunil Kumar is A large number of movies have been made with such themes and a PhD from Mysore characters that would be depicted as ‘scientists’. -

MICROREVIEW Reactivity of Polar Organometallic Compounds In
MICROREVIEW Reactivity of Polar Organometallic Compounds in Unconventional Reaction Media: Challenges and Opportunities Joaquin García-Álvarez,*[a] Eva Hevia,*[b] and Vito Capriati*[c] This paper is gratefully dedicated to the memory of Dr. Guy Lavigne Abstract: Developing new green solvents in designing chemical chemicals in chemical transformations is, indeed, solvents used products and processes or successfully employing the already as reaction media, which account for 80–90% of mass utilization existing ones is one of the key subjects in Green Chemistry and is in a typical pharmaceutical/fine chemical operational process. especially important in Organometallic Chemistry, which is an Thus, the solvent itself is often a critical parameter especially in interdisciplinary field. Can we advantageously use unconventional drug product manufacturing and is as well responsible for most reaction media in place of current harsh organic solvents also for waste generated in the chemical industries and laboratories.[3] polar organometallic compounds? This Microreview critically Following these considerations, some of the most critical analyses the state-of-the-art on this topic and showcases recent and intriguing questions that arise are: Can we get traditional developments and breakthroughs which are becoming new research organic solvents out of organometallic reactions?[4] Can we use directions in this field. Because metals cover a vast swath of the protic, recyclable, biodegradable, and cheap unconventional periodic table, the content is organised into three Sections solvents also for highly reactive organometallic compounds? discussing the reactivity of organometallic compounds of s-, p- and Answering these questions would not only mean to break new d-block elements in unconventional solvents.