Helena Sanches Marcon Caracterização De
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Trees for Farm Forestry: 22 Promising Species
Forestry and Forest Products Natural Heritage Trust Helping Communities Helping Australia TREES FOR FARM FORESTRY: 22 PROMISING SPECIES Forestry and Forest Products TREES FOR FARM FORESTRY: Natural Heritage 22 PROMISING SPECIES Trust Helping Communities Helping Australia A report for the RIRDC/ Land & Water Australia/ FWPRDC Joint Venture Agroforestry Program Revised and Edited by Bronwyn Clarke, Ian McLeod and Tim Vercoe March 2009 i © 2008 Rural Industries Research and Development Corporation. All rights reserved. ISBN 1 74151 821 0 ISSN 1440-6845 Trees for Farm Forestry: 22 promising species Publication No. 09/015 Project No. CSF-56A The information contained in this publication is intended for general use to assist public knowledge and discussion and to help improve the development of sustainable regions. You must not rely on any information contained in this publication without taking specialist advice relevant to your particular circumstances. While reasonable care has been taken in preparing this publication to ensure that information is true and correct, the Commonwealth of Australia gives no assurance as to the accuracy of any information in this publication. The Commonwealth of Australia, the Rural Industries Research and Development Corporation (RIRDC), the authors or contributors expressly disclaim, to the maximum extent permitted by law, all responsibility and liability to any person, arising directly or indirectly from any act or omission, or for any consequences of any such act or omission, made in reliance on the contents of this publication, whether or not caused by any negligence on the part of the Commonwealth of Australia, RIRDC, the authors or contributors. The Commonwealth of Australia does not necessarily endorse the views in this publication. -

Chemical Composition and Insecticidal Activities of Essential Oils of Myrtaceae Against Tribolium Castaneum (Coleoptera: Tenebrionidae)
Pol. J. Environ. Stud. Vol. 26, No. 4 (2017), 1653-1662 DOI: 10.15244/pjoes/73800 Original Research Chemical Composition and Insecticidal Activities of Essential Oils of Myrtaceae against Tribolium castaneum (Coleoptera: Tenebrionidae) Saima Siddique1*, Zahida Parveen3, Firdaus-e-Bareen2, Abida Butt4, Muhammad Nawaz Chaudhary1, Muhammad Akram5 1College of Earth and Environmental Sciences, University of the Punjab, 54890-Lahore, Pakistan 2Department of Botany, University of Punjab, Lahore-54890, Pakistan 3Applied Chemistry Research Centre, PCSIR Laboratories Complex, Lahore-54600, Pakistan 4Department of Zoology, University of the Punjab, 54890-Lahore, Pakistan 5Medicinal Botanic Centre, PCSIR Laboratories Complex, Peshawar-25000, Pakistan Received: 22 April 2017 Accepted: 15 May 2017 Abstract The present study was designed to determine chemical composition of essential oils extracted from different species of the Myrtaceae family and to evaluate their insecticidal activities against Tribolium castaneum (Coleoptera: Tenebrionidae). The essential oils of 10 species were extracted by hydrodistillation and analyzed by a gas chromatography-flame ionization detector (GC-FID) and gas chromatography-mass spectrometry (GC-MS). The main component of Eucalyptus crebra, E. microtheca, E. rudis and Melaleuca quinquenervia essential oils was 1,8-cineole (31.6-49.7%). E. melanophloia and E. tereticornis contained p-cymene (41.8-58.1%) as a major component, while Eucalyptus kitsoniana and E. pruinosa essential oils were dominated by α-pinene (25.8-31.4%). Eugenol methyl ether was identified as a major component in M. bracteata essential oil (82.3%). α-Pinene (31.4%) was the main component in the C. viminalis essential oil. Essential oils of all selected plant species showed good insecticidal activities against T. -

Indicative Planting Lists for the Camden LGA
Indicative Planting Lists for the Camden LGA This list is to offer an indicative guide for trees, shrubs and grasses which do well in the Camden Local Government Area . This list is not exclusive but can be used as a helpful guide . Definitions (For the purpose of the Camden LEP) “tree” means any plant with a sturdy, dominant single main stem and (a) is more than 3 metres high or (b) has a spread of more than 3 metres or (c) has a trunk diameter of more that 150mm measured 1 metre above ground level. “littoral” means the foreshores, riverbanks and the plants of that habitat. “macrophytes” means the conspicuous plants that dominate wetlands, shallow lakes and streams. “salinity” means common salt which is toxic to most land plants when present in high levels in the soil. The selection of street trees should have regard to the following: • Power/Gas/Water/Sewer/Cable Lines • Street Lights • Pruning and shaping resilience of trees • Easements • Driveways & Bus Stops • Pedestrian crossings • House Frontages & Set Backs • Lateral spreading habits of trees • Road Verge & Nature Strip widths • Waste Service collections • Vehicle vision lines • Cultural and Heritage amenity. • Above ground Services. 1 Indicative Nature Strip - Street Tree selection Species Name Common Name Height Width Native Acer palmatum ‘Senkaki’ Coral Bark Maple 4m 3m Acer rubrum ‘October Red Maple 9m 7m Glory’ Acmena smithii ‘Red Red Head Acmena 6m 2m yes Head’ Agonis flexuosa Willow Myrtle 8m 4m yes Angophora costata Dwarf Dwarf Angophora 4m 2m yes ‘Darni’ costata ‘Darni’ -

Fungal Planet Description Sheets: 320–370
Persoonia 34, 2015: 167–266 www.ingentaconnect.com/content/nhn/pimj RESEARCH ARTICLE http://dx.doi.org/10.3767/003158515X688433 Fungal Planet description sheets: 320–370 P.W. Crous1,2,3, M.J. Wingfield2, J. Guarro 4, M. Hernández-Restrepo1,2, D.A. Sutton5, K. Acharya6, P.A. Barber7,. T Boekhout1, R.A. Dimitrov8, M. Dueñas9, A.K. Dutta6, J. Gené4, D.E. Gouliamova10, M. Groenewald1, L. Lombard1, O.V. Morozova11,12, J. Sarkar 6, M.Th. Smith1, A.M. Stchigel4, N.P. Wiederhold 5,. A.V Alexandrova11,13, I. Antelmi14, J. Armengol15, I. Barnes16, J.F. Cano-Lira 4, R.F. Castañeda Ruiz17, M. Contu18, Pr.R. Courtecuisse19, A.L. da Silveira20, C.A. Decock21, A. de Goes20, J. Edathodu22, E. Ercole23, A.C. Firmino20, A. Fourie16, J. Fournier 24, E.L. Furtado25, A.D.W. Geering26, J. Gershenzon27, A. Giraldo4, D. Gramaje28, A. Hammerbacher27, X.-L. He29, D. Haryadi 30, W. Khemmuk26, A.E. Kovalenko11,12, R. Krawczynski31, F. Laich32, C. Lechat33, U.P. Lopes34, H. Madrid35, E.F. Malysheva12,. Y Marín-Felix4, M.P. Martín9, L. Mostert 36, F. Nigro14, O.L. Pereira34, B. Picillo37, D.B. Pinho34, E.S. Popov11,12, C.A. Rodas Peláez 38, S. Rooney-Latham39, M. Sandoval-Denis 4, R.G. Shivas40, V. Silva35, M.M. Stoilova-Disheva10, M.T. Telleria9, C. Ullah 27, S.B. Unsicker 27, N.A. van der Merwe16, A. Vizzini 23, H.-G. Wagner 41, P.T.W. Wong 42, A.R. Wood 43, J.Z. Groenewald1 Key words Abstract Novel species of fungi described in the present study include the following from Malaysia: Castanediella eucalypti from Eucalyptus pellita, Codinaea acacia from Acacia mangium, Emarcea eucalyptigena from Eucalyptus ITS DNA barcodes brassiana, Myrtapenidiella eucalyptorum from Eucalyptus pellita, Pilidiella eucalyptigena from Eucalyptus brassiana LSU and Strelitziana malaysiana from Acacia mangium. -

Guava (Eucalyptus) Rust Puccinia Psidii
INDUSTRY BIOSECURITY PLAN FOR THE NURSERY & GARDEN INDUSTRY Threat Specific Contingency Plan Guava (eucalyptus) rust Puccinia psidii Plant Health Australia March 2009 Disclaimer The scientific and technical content of this document is current to the date published and all efforts were made to obtain relevant and published information on the pest. New information will be included as it becomes available, or when the document is reviewed. The material contained in this publication is produced for general information only. It is not intended as professional advice on any particular matter. No person should act or fail to act on the basis of any material contained in this publication without first obtaining specific, independent professional advice. Plant Health Australia and all persons acting for Plant Health Australia in preparing this publication, expressly disclaim all and any liability to any persons in respect of anything done by any such person in reliance, whether in whole or in part, on this publication. The views expressed in this publication are not necessarily those of Plant Health Australia. Further information For further information regarding this contingency plan, contact Plant Health Australia through the details below. Address: Suite 5, FECCA House 4 Phipps Close DEAKIN ACT 2600 Phone: +61 2 6215 7700 Fax: +61 2 6260 4321 Email: [email protected] Website: www.planthealthaustralia.com.au PHA & NGIA | Contingency Plan – Guava rust (Puccinia psidii) 1 Purpose and background of this contingency plan ............................................................. -

The Flower Chain the Early Discovery of Australian Plants
The Flower Chain The early discovery of Australian plants Hamilton and Brandon, Jill Douglas Hamilton Duchess of University of Sydney Library Sydney, Australia 2002 http://setis.library.usyd.edu.au/ozlit © University of Sydney Library. The texts and images are not to be used for commercial purposes without permission Source Text: Prepared with the author's permission from the print edition published by Kangaroo Press Sydney 1998 All quotation marks are retained as data. First Published: 1990 580.994 1 Australian Etext Collections at botany prose nonfiction 1940- women writers The flower chain the early discovery of Australian plants Sydney Kangaroo Press 1998 Preface Viewing Australia through the early European discovery, naming and appreciation of its flora, gives a fresh perspective on the first white people who went to the continent. There have been books on the battle to transform the wilderness into an agriculturally ordered land, on the convicts, on the goldrush, on the discovery of the wealth of the continent, on most aspects of settlement, but this is the first to link the story of the discovery of the continent with the slow awareness of its unique trees, shrubs and flowers of Australia. The Flower Chain Chapter 1 The Flower Chain Begins Convict chains are associated with early British settlement of Australia, but there were also lighter chains in those grim days. Chains of flowers and seeds to be grown and classified stretched across the oceans from Botany Bay to Europe, looping back again with plants and seeds of the old world that were to Europeanise the landscape and transform it forever. -

A Review of Literature on Ironbark Ridges and Associated Lands ~ Barry Craze ~
A REVIEW OF LITERATURE ON IRONBARK RIDGES AND ASSOCIATED LANDS by Barry Craze and Jim Salmon 2004 FOREWORD This report is dedicated to the memory of Jim Salmon and Barry Craze. It is the last publication that they both researched and wrote before they became ill. Jim and Barry devoted a great deal of their careers to the conservation of soil and the natural environment, and through this report they pass on the wealth of knowledge they both possessed on soils and the management of ironbark country in New South Wales. The text in this book is unchanged from what Jim and Barry originally compiled by the late 1990s. However, most of their original photos that were to be used in the book were lost and have been replaced by new ones. ~ Jim Salmon ~ Jim Salmon was born in Echuca on 30 May 1952 and moved with his parents to Wagga at the age of five. After schooling in Wagga Wagga, he attended Wagga Agricultural College where he graduated with a Diploma of Agriculture. Jim started work with the Soil Conservation Service on 2 June 1974 at Parkes with Kevin McPhee, who was then the District Soil Conservationist. After training with Kevin, Jim was transferred to the job of District Soil Conservationist in Temora on 18 July 1977. It was at Temora that Jim met Helen Hawkins. They married in January 1980 and his three sons David, Scott and Jason were born there in the five years that followed. In September 1989, he took up a property planning position at Temora which he did until May 1993, when he transferred to the Temora District Manager position after David Priem moved to Gundagai. -
Eton Range Realignment Project ATTACHMENT 2 to EPBC Ref: 2015/7552 Preliminary Documentation Residual Impact Assessment and Offset Proposal - 37
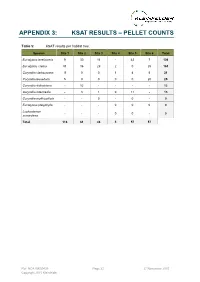
APPENDIX 3: KSAT RESULTS – PELLET COUNTS Table 5: KSAT results per habitat tree. Species Site 1 Site 2 Site 3 Site 4 Site 5 Site 6 Total Eucalyptus tereticornis 9 30 16 - 42 7 104 Eucalyptus crebra 91 16 29 2 0 25 163 Corymbia clarksoniana 11 0 0 1 4 5 21 Corymbia tessellaris 5 0 0 0 0 20 25 Corymbia dallachiana - 12 - - - - 12 Corymbia intermedia - 3 1 0 11 - 15 Corymbia erythrophloia - - 0 - 0 - 0 Eucalyptus platyphylla - - - 0 0 0 0 Lophostemon - - - 0 0 - 0 suaveolens Total 116 61 46 3 57 57 Ref: NCA15R30439 Page 22 27 November 2015 Copyright 2015 Kleinfelder APPENDIX 4: SITE PHOTOS The following images were taken from the centre of each BioCondition quadrat and represent a north east south west aspect, top left to bottom right. Ref: NCA15R30439 Page 23 27 November 2015 Copyright 2015 Kleinfelder Plate 3: BioCondition quadrat 1 (RE11.3.4/11.12.3) Ref: NCA15R30439 Page 24 27 November 2015 Copyright 2015 Kleinfelder Plate 4: BioCondition quadrat 2 (RE11.3.4/11.12.3) Ref: NCA15R30439 Page 25 27 November 2015 Copyright 2015 Kleinfelder Plate 5: BioCondition quadrat 3 (RE11.12.3) Ref: NCA15R30439 Page 26 27 November 2015 Copyright 2015 Kleinfelder Plate 6: BioCondition quadrat 4 (RE11.3.9) Ref: NCA15R30439 Page 27 27 November 2015 Copyright 2015 Kleinfelder Plate 7: BioCondition quadrat 5 (RE11.3.25) Ref: NCA15R30439 Page 28 27 November 2015 Copyright 2015 Kleinfelder Plate 8: BioCondition quadrat 6 (RE11.12.3/11.3.4/11.3.9) Ref: NCA15R30439 Page 29 27 November 2015 Copyright 2015 Kleinfelder Appendix E: Desktop Assessment for Potential -

Eucalyptus Crebra F.Muell
Australian Tropical Rainforest Plants - Online edition Eucalyptus crebra F.Muell. Family: Myrtaceae Mueller, F.J.H. von (1859) Journal Linnean Society, Botany 3: 87. Type: Queensland, Burdekin, 10 Nov. 1856,F. Mueller; holo: MEL. Fide S. T. Blake (1953) Aust. J. Bot. 1: 304. Common name: Red Ironbark; White Ironbark; Narrow Leaved Ironbark; Narrow Leaved Red Ironbark; Ironbark, White; Ironbark, Red; Ironbark, Narrow Leaved; Ironbark, Grey; Grey Ironbark Stem Dead bark dark, almost black on the outer surface; layered when viewed in transverse section, the layers being full of gum veins. Buds and flowers. © R.L. Barrett Leaves Oil dots somewhat scattered. Intramarginal vein very close to the margin and almost indistinguishable from it. Leaf blades about 6-13 x 0.7-1.5 cm. Petioles yellowish. Crushed leaves emit a strong eucalyptus oil odour. Flowers Individual flowers pedicellate. Operculum conical to hemispherical, about 2-3 mm diam., shorter than the calyx tube (hypanthium). Outer operculum shed early, well before the mature bud stage. Fruit Fruits pedicellate, hemispherical or ovoid, about 4-7 x 4-6 mm. Valves 3-4, +/- level with the apex or Scale bar 10mm. © CSIRO included. Seedlings Cotyledons about 3-5 x 5-8 mm, oil dots visible along the margins, petioles about 3-5 mm long. First leaf blades about 20-27 x 4-5 mm, petioles about 3-4 mm long. At the tenth leaf stage: leaf blades about 40-80 x 6-8 mm. petiole about 206 mm long. Oil dots visible in all parts of the leaf blade with the aid of a lens. -

Eucalyptus Crebra Click on Images to Enlarge
Species information Abo ut Reso urces Hom e A B C D E F G H I J K L M N O P Q R S T U V W X Y Z Eucalyptus crebra Click on images to enlarge Family Myrtaceae Scientific Name Eucalyptus crebra F.Muell. Mueller, F.J.H. von (1859) Journal Linnean Society, Botany 3: 87. Type: Queensland, Burdekin, 10 Nov. 1856,F. Buds and flowers. Copyright R.L. Barrett Mueller; holo: MEL. Fide S. T. Blake (1953) Aust. J. Bot. 1: 304. Common name Red Ironbark; White Ironbark; Narrow Leaved Ironbark; Narrow Leaved Red Ironbark; Ironbark, White; Ironbark, Red; Ironbark, Narrow Leaved; Ironbark, Grey; Grey Ironbark Stem Dead bark dark, almost black on the outer surface; layered when viewed in transverse section, the layers being full of gum veins. Scale bar 10mm. Copyright CSIRO Leaves Oil dots somewhat scattered. Intramarginal vein very close to the margin and almost indistinguishable from it. Leaf blades about 6-13 x 0.7-1.5 cm. Petioles yellowish. Crushed leaves emit a strong eucalyptus oil odour. Flowers Individual flowers pedicellate. Operculum conical to hemispherical, about 2-3 mm diam., shorter than the calyx tube (hypanthium). Outer operculum shed early, well before the mature bud stage. Fruit Fruits pedicellate, hemispherical or ovoid, about 4-7 x 4-6 mm. Valves 3-4, +/- level with the apex or included. Cotyledon stage, epigeal germination. Copyright CSIRO Seedlings Cotyledons about 3-5 x 5-8 mm, oil dots visible along the margins, petioles about 3-5 mm long. First leaf blades about 20-27 x 4-5 mm, petioles about 3-4 mm long. -

Eucalypt Open Woodlands
NVIS Fact sheet MVG 11 – Eucalypt open woodlands Australia’s native vegetation is a rich and fundamental Overview element of our natural heritage. It binds and nourishes our ancient soils; shelters and sustains wildlife, protects Typically, vegetation areas classified under MVG 11 – streams, wetlands, estuaries, and coastlines; and absorbs Eucalypt open woodlands: carbon dioxide while emitting oxygen. The National • are characterised by broad spacing with <20 per cent Vegetation Information System (NVIS) has been developed crown cover (<10 per cent foliage projective cover) and maintained by all Australian governments to provide between canopy trees often resulting in a parkland a national picture that captures and explains the broad appearance with a prominent ground layer diversity of our native vegetation. • comprise overstoreys dominated by drought-tolerant eucalypts close to the arid limits of trees This is part of a series of fact sheets which the Australian Government developed based on NVIS Version 4.2 data to • feature understoreys characterised by xeromorphic plants provide detailed descriptions of the major vegetation groups that vary between depositional and upland landforms (MVGs) and other MVG types. The series is comprised of • occur in a central band across semi-arid Australia from a fact sheet for each of the 25 MVGs to inform their use by Western Australia to western New South Wales planners and policy makers. An additional eight MVGs are • are replaced by MVGs 5 and 12 in more humid climates available outlining other MVG types. • are prone to occasional fires in upland areas For more information on these fact sheets, including • are used primarily for broad-scale pastoralism. -

D.Nicolle, Classification of the Eucalypts (Angophora, Corymbia and Eucalyptus) | 2
Taxonomy Genus (common name, if any) Subgenus (common name, if any) Section (common name, if any) Series (common name, if any) Subseries (common name, if any) Species (common name, if any) Subspecies (common name, if any) ? = Dubious or poorly-understood taxon requiring further investigation [ ] = Hybrid or intergrade taxon (only recently-described and well-known hybrid names are listed) ms = Unpublished manuscript name Natural distribution (states listed in order from most to least common) WA Western Australia NT Northern Territory SA South Australia Qld Queensland NSW New South Wales Vic Victoria Tas Tasmania PNG Papua New Guinea (including New Britain) Indo Indonesia TL Timor-Leste Phil Philippines ? = Dubious or unverified records Research O Observed in the wild by D.Nicolle. C Herbarium specimens Collected in wild by D.Nicolle. G(#) Growing at Currency Creek Arboretum (number of different populations grown). G(#)m Reproductively mature at Currency Creek Arboretum. – (#) Has been grown at CCA, but the taxon is no longer alive. – (#)m At least one population has been grown to maturity at CCA, but the taxon is no longer alive. Synonyms (commonly-known and recently-named synonyms only) Taxon name ? = Indicates possible synonym/dubious taxon D.Nicolle, Classification of the eucalypts (Angophora, Corymbia and Eucalyptus) | 2 Angophora (apples) E. subg. Angophora ser. ‘Costatitae’ ms (smooth-barked apples) A. subser. Costatitae, E. ser. Costatitae Angophora costata subsp. euryphylla (Wollemi apple) NSW O C G(2)m A. euryphylla, E. euryphylla subsp. costata (smooth-barked apple, rusty gum) NSW,Qld O C G(2)m E. apocynifolia Angophora leiocarpa (smooth-barked apple) Qld,NSW O C G(1) A.