(SLC12A6) Promoter Polymorphism Leading to an Additional DNA Methylation Site
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

NBD-17-173R1 Title
Elsevier Editorial System(tm) for Neurobiology of Disease Manuscript Draft Manuscript Number: NBD-17-173R1 Title: KCC3 loss-of-function contributes to Andermann syndrome by inducing activity-dependent neuromuscular junction defects Article Type: Research paper Keywords: Motoneuron; Andermann syndrome; chloride homeostasis; electrical activity; neuromuscular junction; Na+/K+ ATPase Corresponding Author: Dr frederique scamps, Corresponding Author's Institution: Inserm First Author: Mélissa Bowerman Order of Authors: Mélissa Bowerman; Céline Salsac; Véronique Bernard; Claire Soulard; Annie Dionne; Emmanuelle Coque; Salim Benlefki; Pascale Hince; Patrick Dion; Gillian Butler-Browne; William Camu; Jean-Pierre Bouchard; Eric Delpire; Guy Rouleau; Cédric Raoul; frederique scamps Abstract: Loss-of-function mutations in the potassium-chloride cotransporter KCC3 lead to Andermann syndrome, a severe sensorimotor neuropathy characterized by areflexia, amyotrophy and locomotor abnormalities. The molecular events responsible for axonal loss remain poorly understood. Here, we establish that global or neuron-specific KCC3 loss-of-function in mice leads to early neuromuscular junction (NMJ) abnormalities and muscular atrophy that are consistent with the pre- synaptic neurotransmission defects observed in patients. KCC3 depletion does not modify chloride handling, but promotes an abnormal electrical activity among primary motoneurons and mislocalization of Na+/K+-ATPase α1 in spinal cord motoneurons. Moreover, the activity-targeting drug carbamazepine restores Na+/K+-ATPase α1 localization and reduces NMJ denervation in Slc12a6-/- mice. We here propose that abnormal motoneuron electrical activity contributes to the peripheral neuropathy observed in Andermann syndrome. Cover Letter Dear Editor Thank you for your interest in this study. We have answered to all the points addressed by the Referees. We now provide discussion and references concerning the pertinence of using motoneuron primary culture relative to recordings from post-natal or adult spinal cord slices. -

Peripheral Neuropathy in Complex Inherited Diseases: an Approach To
PERIPHERAL NEUROPATHY IN COMPLEX INHERITED DISEASES: AN APPROACH TO DIAGNOSIS Rossor AM1*, Carr AS1*, Devine H1, Chandrashekar H2, Pelayo-Negro AL1, Pareyson D3, Shy ME4, Scherer SS5, Reilly MM1. 1. MRC Centre for Neuromuscular Diseases, UCL Institute of Neurology and National Hospital for Neurology and Neurosurgery, London, WC1N 3BG, UK. 2. Lysholm Department of Neuroradiology, National Hospital for Neurology and Neurosurgery, London, WC1N 3BG, UK. 3. Unit of Neurological Rare Diseases of Adulthood, Carlo Besta Neurological Institute IRCCS Foundation, Milan, Italy. 4. Department of Neurology, University of Iowa, 200 Hawkins Drive, Iowa City, IA 52242, USA 5. Department of Neurology, University of Pennsylvania, Philadelphia, PA 19014, USA. * These authors contributed equally to this work Corresponding author: Mary M Reilly Address: MRC Centre for Neuromuscular Diseases, 8-11 Queen Square, London, WC1N 3BG, UK. Email: [email protected] Telephone: 0044 (0) 203 456 7890 Word count: 4825 ABSTRACT Peripheral neuropathy is a common finding in patients with complex inherited neurological diseases and may be subclinical or a major component of the phenotype. This review aims to provide a clinical approach to the diagnosis of this complex group of patients by addressing key questions including the predominant neurological syndrome associated with the neuropathy e.g. spasticity, the type of neuropathy, and the other neurological and non- neurological features of the syndrome. Priority is given to the diagnosis of treatable conditions. Using this approach, we associated neuropathy with one of three major syndromic categories - 1) ataxia, 2) spasticity, and 3) global neurodevelopmental impairment. Syndromes that do not fall easily into one of these three categories can be grouped according to the predominant system involved in addition to the neuropathy e.g. -
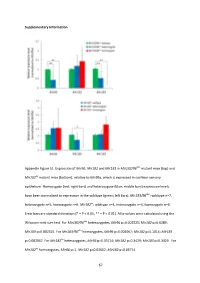
And Mir183 in Mir183/96 Dko Mutant Mice (Top) And
Supplementary Information Appendix Figure S1. Expression of Mir96 , Mir182 and Mir183 in Mir183/96 dko mutant mice (top) and Mir182 ko mutant mice (bottom), relative to Mir99a , which is expressed in cochlear sensory epithelium. Homozygote (red; right bars) and heterozygote (blue; middle bars) expression levels have been normalised to expression in the wildtype (green; left bars). Mir183/96 dko : wildtype n=7, heterozygote n=5, homozygote n=6. Mir182 ko : wildtype n=4, heterozygote n=4, homozygote n=4. Error bars are standard deviation (* = P < 0.05, ** = P < 0.01). All p-values were calculated using the Wilcoxon rank sum test. For Mir183/96 dko heterozygotes, Mir96 p=0.002525; Mir182 p=0.6389; Mir183 p=0.002525. For Mir183/96 dko homozygotes, Mir96 p=0.002067; Mir182 p=0.1014; Mir183 p=0.002067. For Mir182 ko heterozygotes, Mir96 p=0.05714; Mir182 p=0.3429; Mir183 p=0.3429. For Mir182 ko homozygotes, Mir96 p=1; Mir182 p=0.02652; Mir183 p=0.05714. 67 68 Appendix Figure S2. Individual ABR thresholds of wildtype, heterozygous and homozygous Mir183/96 dko mice at all ages tested. Number of mice of each genotype tested at each age is shown on the threshold plot. 69 70 Appendix Figure S3. Individual ABR thresholds of wildtype, heterozygous and homozygous Mir182 ko mice at all ages tested. Number of mice of each genotype tested at each age is shown on the threshold plot. 71 Appendix Figure S4. Mean ABR waveforms at 12kHz, shown at 20dB (top) and 50dB (bottom) above threshold (sensation level, SL) ± standard deviation, at four weeks old. -

The Genetic Landscape of the Human Solute Carrier (SLC) Transporter Superfamily
Human Genetics (2019) 138:1359–1377 https://doi.org/10.1007/s00439-019-02081-x ORIGINAL INVESTIGATION The genetic landscape of the human solute carrier (SLC) transporter superfamily Lena Schaller1 · Volker M. Lauschke1 Received: 4 August 2019 / Accepted: 26 October 2019 / Published online: 2 November 2019 © The Author(s) 2019 Abstract The human solute carrier (SLC) superfamily of transporters is comprised of over 400 membrane-bound proteins, and plays essential roles in a multitude of physiological and pharmacological processes. In addition, perturbation of SLC transporter function underlies numerous human diseases, which renders SLC transporters attractive drug targets. Common genetic polymorphisms in SLC genes have been associated with inter-individual diferences in drug efcacy and toxicity. However, despite their tremendous clinical relevance, epidemiological data of these variants are mostly derived from heterogeneous cohorts of small sample size and the genetic SLC landscape beyond these common variants has not been comprehensively assessed. In this study, we analyzed Next-Generation Sequencing data from 141,456 individuals from seven major human populations to evaluate genetic variability, its functional consequences, and ethnogeographic patterns across the entire SLC superfamily of transporters. Importantly, of the 204,287 exonic single-nucleotide variants (SNVs) which we identifed, 99.8% were present in less than 1% of analyzed alleles. Comprehensive computational analyses using 13 partially orthogonal algorithms that predict the functional impact of genetic variations based on sequence information, evolutionary conserva- tion, structural considerations, and functional genomics data revealed that each individual genome harbors 29.7 variants with putative functional efects, of which rare variants account for 18%. Inter-ethnic variability was found to be extensive, and 83% of deleterious SLC variants were only identifed in a single population. -

Nephritis Responses in Murine and Human Lupus Analysis Defines
Downloaded from http://www.jimmunol.org/ by guest on October 2, 2021 is online at: average * The Journal of Immunology , 24 of which you can access for free at: 2012; 189:988-1001; Prepublished online 20 June from submission to initial decision 4 weeks from acceptance to publication Celine C. Berthier, Ramalingam Bethunaickan, Tania Gonzalez-Rivera, Viji Nair, Meera Ramanujam, Weijia Zhang, Erwin P. Bottinger, Stephan Segerer, Maja Lindenmeyer, Clemens D. Cohen, Anne Davidson and Matthias Kretzler 2012; doi: 10.4049/jimmunol.1103031 http://www.jimmunol.org/content/189/2/988 Cross-Species Transcriptional Network Analysis Defines Shared Inflammatory Responses in Murine and Human Lupus Nephritis J Immunol cites 60 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://www.jimmunol.org/content/189/2/988.full#ref-list-1 http://www.jimmunol.org/content/suppl/2012/06/20/jimmunol.110303 1.DC1 This article Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2012 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of October 2, 2021. The Journal of Immunology Cross-Species Transcriptional Network Analysis Defines Shared Inflammatory Responses in Murine and Human Lupus Nephritis Celine C. -

The Genetics of Bipolar Disorder
Molecular Psychiatry (2008) 13, 742–771 & 2008 Nature Publishing Group All rights reserved 1359-4184/08 $30.00 www.nature.com/mp FEATURE REVIEW The genetics of bipolar disorder: genome ‘hot regions,’ genes, new potential candidates and future directions A Serretti and L Mandelli Institute of Psychiatry, University of Bologna, Bologna, Italy Bipolar disorder (BP) is a complex disorder caused by a number of liability genes interacting with the environment. In recent years, a large number of linkage and association studies have been conducted producing an extremely large number of findings often not replicated or partially replicated. Further, results from linkage and association studies are not always easily comparable. Unfortunately, at present a comprehensive coverage of available evidence is still lacking. In the present paper, we summarized results obtained from both linkage and association studies in BP. Further, we indicated new potential interesting genes, located in genome ‘hot regions’ for BP and being expressed in the brain. We reviewed published studies on the subject till December 2007. We precisely localized regions where positive linkage has been found, by the NCBI Map viewer (http://www.ncbi.nlm.nih.gov/mapview/); further, we identified genes located in interesting areas and expressed in the brain, by the Entrez gene, Unigene databases (http://www.ncbi.nlm.nih.gov/entrez/) and Human Protein Reference Database (http://www.hprd.org); these genes could be of interest in future investigations. The review of association studies gave interesting results, as a number of genes seem to be definitively involved in BP, such as SLC6A4, TPH2, DRD4, SLC6A3, DAOA, DTNBP1, NRG1, DISC1 and BDNF. -

SLC12A6 Gene Solute Carrier Family 12 Member 6
SLC12A6 gene solute carrier family 12 member 6 Normal Function The SLC12A6 gene provides instructions for making a protein called a K-Cl cotransporter. This protein is involved in moving charged atoms (ions) of potassium (K) and chlorine (Cl) across the cell membrane. The positively charged potassium ions and negatively charged chlorine ions are moved together (cotransported), so that the charges inside and outside the cell membrane are unchanged (electroneutral). Electroneutral cotransport of ions across cell membranes is involved in many functions of the body. While the specific function of the K-Cl cotransporter produced from the SLC12A6 gene is unknown, it seems to be critical for the development and maintenance of nerve tissue. It may be involved in regulating the amounts of potassium, chlorine, or water in cells and intercellular spaces. The K-Cl cotransporter protein may also help regulate the activity of other proteins that are sensitive to ion concentrations. Health Conditions Related to Genetic Changes Andermann syndrome At least six SLC12A6 gene mutations have been identified in people with Andermann syndrome. Almost all affected individuals of French-Canadian descent have the same mutation in both copies of the SLC12A6 gene, in which the DNA building block ( nucleotide) guanine is deleted at position 2436 (written as 2436delG). This mutation is common in the populations of the Saguenay-Lac-St.-Jean and Charlevoix regions of northeastern Quebec. Most SLC12A6 gene mutations that cause Andermann syndrome result in a K-Cl cotransporter protein that is shortened and nonfunctional. The lack of functional protein produced from the SLC12A6 gene is believed to interfere with the development of the corpus callosum and maintenance of the nerves that transmit signals needed for movement and sensation, resulting in the signs and symptoms of Andermann syndrome. -

Supplementary Table 1
Supplementary Table 1. 492 genes are unique to 0 h post-heat timepoint. The name, p-value, fold change, location and family of each gene are indicated. Genes were filtered for an absolute value log2 ration 1.5 and a significance value of p ≤ 0.05. Symbol p-value Log Gene Name Location Family Ratio ABCA13 1.87E-02 3.292 ATP-binding cassette, sub-family unknown transporter A (ABC1), member 13 ABCB1 1.93E-02 −1.819 ATP-binding cassette, sub-family Plasma transporter B (MDR/TAP), member 1 Membrane ABCC3 2.83E-02 2.016 ATP-binding cassette, sub-family Plasma transporter C (CFTR/MRP), member 3 Membrane ABHD6 7.79E-03 −2.717 abhydrolase domain containing 6 Cytoplasm enzyme ACAT1 4.10E-02 3.009 acetyl-CoA acetyltransferase 1 Cytoplasm enzyme ACBD4 2.66E-03 1.722 acyl-CoA binding domain unknown other containing 4 ACSL5 1.86E-02 −2.876 acyl-CoA synthetase long-chain Cytoplasm enzyme family member 5 ADAM23 3.33E-02 −3.008 ADAM metallopeptidase domain Plasma peptidase 23 Membrane ADAM29 5.58E-03 3.463 ADAM metallopeptidase domain Plasma peptidase 29 Membrane ADAMTS17 2.67E-04 3.051 ADAM metallopeptidase with Extracellular other thrombospondin type 1 motif, 17 Space ADCYAP1R1 1.20E-02 1.848 adenylate cyclase activating Plasma G-protein polypeptide 1 (pituitary) receptor Membrane coupled type I receptor ADH6 (includes 4.02E-02 −1.845 alcohol dehydrogenase 6 (class Cytoplasm enzyme EG:130) V) AHSA2 1.54E-04 −1.6 AHA1, activator of heat shock unknown other 90kDa protein ATPase homolog 2 (yeast) AK5 3.32E-02 1.658 adenylate kinase 5 Cytoplasm kinase AK7 -

Genetic Background of Ataxia in Children Younger Than 5 Years in Finland E444
Volume 6, Number 4, August 2020 Neurology.org/NG A peer-reviewed clinical and translational neurology open access journal ARTICLE Genetic background of ataxia in children younger than 5 years in Finland e444 ARTICLE Cerebral arteriopathy associated with heterozygous variants in the casitas B-lineage lymphoma gene e448 ARTICLE Somatic SLC35A2 mosaicism correlates with clinical fi ndings in epilepsy brain tissuee460 ARTICLE Synonymous variants associated with Alzheimer disease in multiplex families e450 Academy Officers Neurology® is a registered trademark of the American Academy of Neurology (registration valid in the United States). James C. Stevens, MD, FAAN, President Neurology® Genetics (eISSN 2376-7839) is an open access journal published Orly Avitzur, MD, MBA, FAAN, President Elect online for the American Academy of Neurology, 201 Chicago Avenue, Ann H. Tilton, MD, FAAN, Vice President Minneapolis, MN 55415, by Wolters Kluwer Health, Inc. at 14700 Citicorp Drive, Bldg. 3, Hagerstown, MD 21742. Business offices are located at Two Carlayne E. Jackson, MD, FAAN, Secretary Commerce Square, 2001 Market Street, Philadelphia, PA 19103. Production offices are located at 351 West Camden Street, Baltimore, MD 21201-2436. Janis M. Miyasaki, MD, MEd, FRCPC, FAAN, Treasurer © 2020 American Academy of Neurology. Ralph L. Sacco, MD, MS, FAAN, Past President Neurology® Genetics is an official journal of the American Academy of Neurology. Journal website: Neurology.org/ng, AAN website: AAN.com CEO, American Academy of Neurology Copyright and Permission Information: Please go to the journal website (www.neurology.org/ng) and click the Permissions tab for the relevant Mary E. Post, MBA, CAE article. Alternatively, send an email to [email protected]. -

Supporting Information
Supporting Information Table S1. List of confirmed SLC transporters represented in Canine GeneChip. SLC family Members detected Members not detected SLC1: The high affinity glutamate and neutral amino acid SLC1A1 SLC1A2, SLC1A3, SLC1A6 transporter family SLC2: The facilitative GLUT transporter family SLC2A1, SLC2A8 SLC2A3, SLC2A9 SLC3: The heavy subunits of the heteromeric amino acid SLC3A1 transporters SLC4: The bicarbonate transporter family SLC4A11 SLC4A4, SLC4A8 SLC5: The sodium glucose cotransporter family SLC5A6 SLC5A3, SLC5A10, SLC5A12 SLC6: The sodium- and chloride- dependent SLC6A6, SLC6A12 SLCA18 neurotransmitter transporter family SLC7: The cationic amino acid transporter/glycoprotein- NR associated family SLC8: The Na+/Ca2+ exchanger family SLC8A1 SLC9: The Na+/H+ exchanger family SLC9A1, SLC9A6, SLC9A9 SLC10: The sodium bile salt cotransport family SLC10A2 SLC11: The proton coupled metal ion transporter family NR SLC12: The electroneutral cation-Cl cotransporter family SLC12A3, SLC12A6, SLC12A8 SLC13: The human Na+-sulfate/carboxylate cotransporter SLC13A2 family SLC14: The urea transporter family NR SLC15: The proton oligopeptide cotransporter family SLC15A2, SLC15A4 SLC15A1 SLC16: The monocarboxylate transporter family SLC16A13 SLC16A4 SLC17: The vesicular glutamate transporter family SLC17A3, SLC17A7 SLC18: The vesicular amine transporter family NR SLC19: The folate/thiamine transporter family NR SLC20: The type III Na+-phosphate cotransporter family NR SLC21/SLCO: The organic anion transporting family SLC21A3, SLC21A8, -

2017PNS Final Programme W
2017 PNS Annual Meeting 8-12 July | Sitges, Spain Programme and Abstracts Programme Programme and Abstracts 2017 PNS Annual Meeting 8 – 12 July Sitges, Spain Welcome On behalf of the Peripheral Nerve Society, we are delighted to welcome you to the 2017 Annual Meeting at the Meliá in Sitges, Spain. The PNS Annual Meeting continues to be the premier meeting for cutting-edge innovation and advances in peripheral neuropathy. The 2017 Meeting will provide a mixture of excellent plenary lectures, oral platforms, oral posters, poster sessions, an education course and dedicated symposia organised by special interest groups for Charcot Marie Tooth and related neuropathies (CMTR), the Inflammatory Neuropathy Consortium (INC) and diabetes. There will also be clinical trials update sessions and a hot topic symposium. We look forward to you being part of it. Join us for the Opening Ceremony on Saturday, 8 July, from 18.00 to 20.00, in the Auditorium Hall. The reception will feature complimentary drinks and hors d’oeuvres. Coffee breaks and lunch for registrants will only be provided daily for registrants. Please see the programme for details. Complimentary internet will be provided. Instructions for access can be found on page 11 of the programme. Be sure to attend the Business Meeting on Monday at 13.00 in the Main Auditorium. We will be reviewing Society business, and your input is needed. Monday night, the PNS will be honoring Junior and new Members of the Society with a cocktail reception from 19.00 to 20.00. For those who have registered for this event during the online registration, please join us for the PNS Closing Dinner on Tuesday, 11 July, from 19.00 to 22.00 in the Tramuntana room. -

Andermann Syndrome
Andermann syndrome Description Andermann syndrome is a disorder that damages the nerves used for muscle movement and sensation (motor and sensory neuropathy). Absence (agenesis) or malformation of the tissue connecting the left and right halves of the brain (corpus callosum) also occurs in most people with this disorder. People affected by Andermann syndrome have abnormal or absent reflexes (areflexia) and weak muscle tone (hypotonia). They experience muscle wasting (amyotrophy), severe progressive weakness and loss of sensation in the limbs, and rhythmic shaking ( tremors). They typically begin walking between ages 3 and 4 and lose this ability by their teenage years. As they get older, people with this disorder frequently develop joint deformities called contractures, which restrict the movement of certain joints. Most affected individuals also develop abnormal curvature of the spine (scoliosis), which may require surgery. Andermann syndrome also results in abnormal function of certain cranial nerves, which emerge directly from the brain and extend to various areas of the head and neck. Cranial nerve problems may result in facial muscle weakness, drooping eyelids (ptosis), and difficulty following movements with the eyes (gaze palsy). Individuals with Andermann syndrome usually have intellectual disability, which may be mild to severe, and some experience seizures. They may also develop psychiatric symptoms such as depression, anxiety, agitation, paranoia, and hallucinations, which usually appear in adolescence. Some people with Andermann syndrome have atypical physical features such as widely spaced eyes (ocular hypertelorism); a wide, short skull (brachycephaly); a high arch of the hard palate at the roof of the mouth; a big toe that crosses over the other toes; and partial fusion (syndactyly) of the second and third toes.