Investor Relations : Biomarin
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
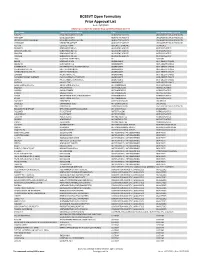
BCBSVT Open Formulary Prior Approval List
BCBSVT Open Formulary Prior Approval List As of: 10/27/2020 Helpful Tip: To search for a specific drug, use the find feature (Ctrl + F) Trade Name Chemical/Biological Name Class Prior Authorization Program FUSILEV LEVOLEUCOVORIN CALCIUM ADJUNCTIVE AGENTS UNCLASSIFIED DRUG PRODUCTS KHAPZORY LEVOLEUCOVORIN ADJUNCTIVE AGENTS UNCLASSIFIED DRUG PRODUCTS LEVOLEUCOVORIN CALCIUM LEVOLEUCOVORIN CALCIUM ADJUNCTIVE AGENTS UNCLASSIFIED DRUG PRODUCTS VISTOGARD URIDINE TRIACETATE ADJUNCTIVE AGENTS UNCLASSIFIED DRUG PRODUCTS ACTHAR CORTICOTROPIN ADRENAL HORMONES HORMONES BELRAPZO BENDAMUSTINE HCL ALKYLATING AGENTS ANTINEOPLASTICS BENDAMUSTINE HCL BENDAMUSTINE HCL ALKYLATING AGENTS ANTINEOPLASTICS BENDEKA BENDAMUSTINE HCL ALKYLATING AGENTS ANTINEOPLASTICS TREANDA BENDAMUSTINE HCL ALKYLATING AGENTS ANTINEOPLASTICS DAW (DISPENSE AS WRITTEN) ALL CUSTOM BELVIQ LORCASERIN HCL ANOREXIANTS ANTI‐OBESITY DRUGS BELVIQ XR LORCASERIN HCL ANOREXIANTS ANTI‐OBESITY DRUGS CONTRAVE ER NALTREXONE HCL/BUPROPION HCL ANOREXIANTS ANTI‐OBESITY DRUGS DIETHYLPROPION HCL DIETHYLPROPION HCL ANOREXIANTS ANTI‐OBESITY DRUGS DIETHYLPROPION HCL ER DIETHYLPROPION HCL ANOREXIANTS ANTI‐OBESITY DRUGS LOMAIRA PHENTERMINE HCL ANOREXIANTS ANTI‐OBESITY DRUGS PHENDIMETRAZINE TARTRATE PHENDIMETRAZINE TARTRATE ANOREXIANTS ANTI‐OBESITY DRUGS QSYMIA PHENTERMINE/TOPIRAMATE ANOREXIANTS ANTI‐OBESITY DRUGS SAXENDA LIRAGLUTIDE ANOREXIANTS ANTI‐OBESITY DRUGS ABIRATERONE ACETATE ABIRATERONE ACETATE ANTIANDROGENS ANTINEOPLASTICS ERLEADA APALUTAMIDE ANTIANDROGENS ANTINEOPLASTICS NUBEQA DAROLUTAMIDE ANTIANDROGENS -

Biomarin Pharmaceutical Inc
BIOMARIN PHARMACEUTICAL INC FORM 10-K (Annual Report) Filed 03/02/15 for the Period Ending 12/31/14 Address 105 DIGITAL DRIVE NOVATO, CA 94949 Telephone 4155066700 CIK 0001048477 Symbol BMRN SIC Code 2834 - Pharmaceutical Preparations Industry Biotechnology & Drugs Sector Healthcare Fiscal Year 12/31 http://www.edgar-online.com © Copyright 2015, EDGAR Online, Inc. All Rights Reserved. Distribution and use of this document restricted under EDGAR Online, Inc. Terms of Use. f UNITED STATES SECURITIES AND EXCHANGE COMMISSION Washington, D.C. 20549 Form 10-K (Mark One) ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 For the fiscal year ended December 31, 2014 Or TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 For the transition period from to . Commission file number: 000-26727 BioMarin Pharmaceutical Inc. (Exact name of registrant as specified in its charter) Delaware 68 -0397820 (State of other jurisdiction of (I.R.S. Employer incorporation or organization) Identification No.) 770 Lindaro Street San Rafael, California 94901 (Address of principal executive offices) (Zip Code) Registrant’s telephone number, including area code: (415) 506-6700 Securities registered pursuant to Section 12(b) of the Act: Title of Each Class Name of Each Exchange on Which Registered Common Stock, $.001 par value The NASDAQ Global Select Market Securities registered under Section 12(g) of the Act: None Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes No Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. -

DRUGS REQUIRING PRIOR AUTHORIZATION in the MEDICAL BENEFIT Page 1
Effective Date: 08/01/2021 DRUGS REQUIRING PRIOR AUTHORIZATION IN THE MEDICAL BENEFIT Page 1 Therapeutic Category Drug Class Trade Name Generic Name HCPCS Procedure Code HCPCS Procedure Code Description Anti-infectives Antiretrovirals, HIV CABENUVA cabotegravir-rilpivirine C9077 Injection, cabotegravir and rilpivirine, 2mg/3mg Antithrombotic Agents von Willebrand Factor-Directed Antibody CABLIVI caplacizumab-yhdp C9047 Injection, caplacizumab-yhdp, 1 mg Cardiology Antilipemic EVKEEZA evinacumab-dgnb C9079 Injection, evinacumab-dgnb, 5 mg Cardiology Hemostatic Agent BERINERT c1 esterase J0597 Injection, C1 esterase inhibitor (human), Berinert, 10 units Cardiology Hemostatic Agent CINRYZE c1 esterase J0598 Injection, C1 esterase inhibitor (human), Cinryze, 10 units Cardiology Hemostatic Agent FIRAZYR icatibant J1744 Injection, icatibant, 1 mg Cardiology Hemostatic Agent HAEGARDA c1 esterase J0599 Injection, C1 esterase inhibitor (human), (Haegarda), 10 units Cardiology Hemostatic Agent ICATIBANT (generic) icatibant J1744 Injection, icatibant, 1 mg Cardiology Hemostatic Agent KALBITOR ecallantide J1290 Injection, ecallantide, 1 mg Cardiology Hemostatic Agent RUCONEST c1 esterase J0596 Injection, C1 esterase inhibitor (recombinant), Ruconest, 10 units Injection, lanadelumab-flyo, 1 mg (code may be used for Medicare when drug administered under Cardiology Hemostatic Agent TAKHZYRO lanadelumab-flyo J0593 direct supervision of a physician, not for use when drug is self-administered) Cardiology Pulmonary Arterial Hypertension EPOPROSTENOL (generic) -

AHFS Pharmacologic-Therapeutic Classification System
AHFS Pharmacologic-Therapeutic Classification System Abacavir 48:24 - Mucolytic Agents - 382638 8:18.08.20 - HIV Nucleoside and Nucleotide Reverse Acitretin 84:92 - Skin and Mucous Membrane Agents, Abaloparatide 68:24.08 - Parathyroid Agents - 317036 Aclidinium Abatacept 12:08.08 - Antimuscarinics/Antispasmodics - 313022 92:36 - Disease-modifying Antirheumatic Drugs - Acrivastine 92:20 - Immunomodulatory Agents - 306003 4:08 - Second Generation Antihistamines - 394040 Abciximab 48:04.08 - Second Generation Antihistamines - 394040 20:12.18 - Platelet-aggregation Inhibitors - 395014 Acyclovir Abemaciclib 8:18.32 - Nucleosides and Nucleotides - 381045 10:00 - Antineoplastic Agents - 317058 84:04.06 - Antivirals - 381036 Abiraterone Adalimumab; -adaz 10:00 - Antineoplastic Agents - 311027 92:36 - Disease-modifying Antirheumatic Drugs - AbobotulinumtoxinA 56:92 - GI Drugs, Miscellaneous - 302046 92:20 - Immunomodulatory Agents - 302046 92:92 - Other Miscellaneous Therapeutic Agents - 12:20.92 - Skeletal Muscle Relaxants, Miscellaneous - Adapalene 84:92 - Skin and Mucous Membrane Agents, Acalabrutinib 10:00 - Antineoplastic Agents - 317059 Adefovir Acamprosate 8:18.32 - Nucleosides and Nucleotides - 302036 28:92 - Central Nervous System Agents, Adenosine 24:04.04.24 - Class IV Antiarrhythmics - 304010 Acarbose Adenovirus Vaccine Live Oral 68:20.02 - alpha-Glucosidase Inhibitors - 396015 80:12 - Vaccines - 315016 Acebutolol Ado-Trastuzumab 24:24 - beta-Adrenergic Blocking Agents - 387003 10:00 - Antineoplastic Agents - 313041 12:16.08.08 - Selective -

Oregon Medicaid Pharmaceutical Services Prior Authorization Criteria
Oregon Medicaid Pharmaceutical Services Prior Authorization Criteria HEALTH SYSTEMS DIVISION Prior authorization (PA) criteria for fee-for-service prescriptions for Oregon Health Plan clients March 1, 2021 Contents Contents ................................................................................................................................................................ 2 Introduction........................................................................................................................................................... 7 About this guide ......................................................................................................................................... 7 How to use this guide ................................................................................................................................. 7 Administrative rules and supplemental information .................................................................................. 7 Update information............................................................................................................................................... 8 Effective March 1, 2021 ............................................................................................................................ 8 Substantive updates and new criteria ............................................................................................. 8 Clerical changes ............................................................................................................................ -

Medicines Not Reimbursed Through National Prices and Directly Commissioned by Nhs England V14 Changes to Version 13
MEDICINES NOT REIMBURSED THROUGH NATIONAL PRICES AND DIRECTLY COMMISSIONED BY NHS ENGLAND V14 CHANGES TO VERSION 13 PUBLISHED APRIL 2019 Changes to v13 Lines removed SUITABLE SPECIALIST FOR CENTRE SUITABLE FOR SHARED ONLY SHARED CARE CARE PRIOR (includng WITH PRIMARY BETWEEN STOPPING MONITORING/ AUDIT APPROVAL outreach CARE (IF DRUG NAME INDICATION COMMISSIONER PBR CATEGORY TA/POLICY STARTING CRITERIA SPECIALIST COMMENT CRITERIA REQUIREMENTS PROFORMA when SUPPORTED AND REQUIRED delivered as BY LOCAL SECONDARY part of a PRESCRIBING CARE VIA provider COMMITTEE) NETWORK network) MODEL PAEDIATRIC INDICATIONS (IN LINE WITH NHS ENGLAND AS PER ADULT TA'S (TA195, TA373, ABATACEPT NHS ENGLAND CYTOKINE MODULATORS NICE NICE NICE √ MEDICINES FOR CHILDREN TA375) POLICY) Now covered by comment 8 PAEDIATRIC INDICATIONS (IN AS PER TA455 OR ADULT TA'S (TA130 LINE WITH NHS ENGLAND (replaced by TA375), TA143 (repalced by ADALIMUMAB NHS ENGLAND CYTOKINE MODULATORS NICE NICE NICE AUDIT √ √ MEDICINES FOR CHILDREN TA383), TA146, TA187, TA199, TA329, POLICY) TA392, TA460) Now covered by comment 8 ALBUTREPENONACOG ALFA HAEMOPHILIA B PRODUCTS ON CMU Blood factor products simplified NHS ENGLAND BLOOD-RELATED PRODUCTS SSC 1652 SSC 1652 SSC 1652 √ FRAMEWORK to blood factor product VIII etc PAEDIATRIC INDICATIONS (IN LINE WITH NHS ENGLAND AS PER IFR AS PER IFR ANAKINRA NHS ENGLAND CYTOKINE MODULATORS NOT ROUTINELY COMMISSIONED AS PER IFR APPROVAL √ MEDICINES FOR CHILDREN APPROVAL APPROVAL POLICY) Now covered by comment 8 AS PER BCSH GUIDELINES ANTIHAEMOPHILIC FACTOR/VON -

Reducing Prescription Drug Costs in Colorado Cost Drivers and Strategies to Address Them
2nd Edition Reducing Prescription Drug Costs in Colorado Cost Drivers and Strategies to Address Them January 2021 Contents Forward to Reducing Prescription Drug Costs in Colorado, 2nd Edition ........... 4 2020 Updates to This Report .................................................................. 4 Executive Summary ............................................................................ 6 Introduction and Purpose ...................................................................... 6 Cost Drivers ...................................................................................... 7 Prioritized Solutions ............................................................................ 8 Learning from Medicaid Policy ............................................................... 12 The Department Invites Your Collaborative Partnership .................................. 12 Industry Trends and Background Information ............................................. 13 Prescription Drug Cost, Utilization, and Trends ........................................... 13 The U.S. Pays the Highest Prices for Pharmaceutical Drugs in the World .............. 16 Many Coloradans Aren’t Taking Their Drugs Appropriately Because They Can’t Afford Them, Often Leading to Worse Health Outcomes That Are More Costly ....... 16 Major Drivers of Prescription Drug Prices .................................................. 18 Patent Protections .............................................................................. 18 Anti-Competitive Practices and Price Fixing -

Pegvaliase-Pqpz
Pharmacy Benefit Coverage Criteria Effective Date………….………….....................12/1/2020 Next Review Date………………………………. 12/1/2021 Coverage Policy Number ................................ P0055 Pegvaliase-pqpz Table of Contents Related Coverage Resources Medical Necessity Criteria ................................... 1 Sapropterin FDA Approved Indications ................................... 2 Recommended Dosing ........................................ 2 General Background ............................................ 3 References .......................................................... 3 INSTRUCTIONS FOR USE The following Coverage Policy applies to health benefit plans administered by Cigna Companies. Certain Cigna Companies and/or lines of business only provide utilization review services to clients and do not make coverage determinations. References to standard benefit plan language and coverage determinations do not apply to those clients. Coverage Policies are intended to provide guidance in interpreting certain standard benefit plans administered by Cigna Companies. Please note, the terms of a customer’s particular benefit plan document [Group Service Agreement, Evidence of Coverage, Certificate of Coverage, Summary Plan Description (SPD) or similar plan document] may differ significantly from the standard benefit plans upon which these Coverage Policies are based. For example, a customer’s benefit plan document may contain a specific exclusion related to a topic addressed in a Coverage Policy. In the event of a conflict, a customer’s -

Specialty Pharmacy Program Drug List
Specialty Pharmacy Program Drug List The Specialty Pharmacy Program covers certain drugs commonly referred to as high-cost Specialty Drugs. To receive in- network benefits/coverage for these drugs, these drugs must be dispensed through a select group of contracted specialty pharmacies or your medical provider. Please call the BCBSLA Customer Service number on the back of your member ID card for information about our contracted specialty pharmacies. All specialty drugs listed below are limited to the retail day supply listed in your benefit plan (typically a 30-day supply). As benefits may vary by group and individual plans, the inclusion of a medication on this list does not imply prescription drug coverage. Please refer to your benefit plan for a complete list of benefits, including specific exclusions, limitations and member cost-sharing amounts you are responsible for such as a deductible, copayment and coinsurance. Brand Name Generic Name Drug Classification 8-MOP methoxsalen Psoralen ACTEMRA SC tocilizumab Monoclonal Antibody/Arthritis ACTHAR corticotropin Adrenocortical Insufficiency ACTIMMUNE interferon gamma 1b Interferon ADCIRCA tadalafil Pulmonary Vasodilator ADEMPAS riociguat Pulmonary Vasodilator AFINITOR everolimus Oncology ALECENSA alectinib Oncology ALKERAN (oral) melphalan Oncology ALUNBRIG brigatinib Oncology AMPYRA ER dalfampridine Multiple Sclerosis APTIVUS tipranavir HIV/AIDS APOKYN apomorphine Parkinson's Disease ARCALYST rilonacept Interleukin Blocker/CAPS ATRIPLA efavirenz-emtricitabine-tenofovir HIV/AIDS AUBAGIO -

PALYNZIQ (Pegvaliase-Pqpz) Injection, for Subcutaneous Use Blood Phenylalanine Monitoring and Diet (2.2) Initial U.S
HIGHLIGHTS OF PRESCRIBING INFORMATION 600 micromol/L after 16 weeks of continuous treatment with the These highlights do not include all the information needed to use maximum dosage of 40 mg once daily. PALYNZIQ safely and effectively. See full prescribing information for Reduce the dosage and/or modify dietary protein and phenylalanine PALYNZIQ. intake, as needed, to maintain blood phenylalanine concentrations within a clinically acceptable range and above 30 micromol/L. PALYNZIQ (pegvaliase-pqpz) injection, for subcutaneous use Blood Phenylalanine Monitoring and Diet (2.2) Initial U.S. Approval: 2018 Obtain blood phenylalanine concentrations every 4 weeks until a maintenance dosage is established. WARNING: RISK OF ANAPHYLAXIS After a maintenance dosage is established, periodically monitor blood See full prescribing information for complete boxed warning. phenylalanine concentrations. Anaphylaxis has been reported after administration of Palynziq Counsel patients to monitor dietary protein and phenylalanine intake, and may occur at any time during treatment. (5.1) and adjust as directed by their healthcare provider. Administer the initial dose of Palynziq under the supervision of a Premedication (2.3, 5.1, 5.3) healthcare provider equipped to manage anaphylaxis, and closely Consider premedication for hypersensitivity reactions. observe patients for at least 60 minutes following injection. Prior Administration Instructions (2.4) to self-injection, confirm patient competency with Rotate injection sites. If more than one injection is needed for a single self-administration, and patient’s and observer’s (if applicable) dose, the injection sites should be at least 2 inches away from each other. ability to recognize signs and symptoms of anaphylaxis and to administer auto-injectable epinephrine, if needed. -

Self-Administered Specialty Drug List
Self‐Administered Drug List BRAND NAME GENERIC DRUG NME ABIRATERONE ACETATE ABIRATERONE ACETATE ACTEMRA TOCILIZUMAB ACTEMRA ACTPEN TOCILIZUMAB ACTHAR CORTICOTROPIN ACTIMMUNE INTERFERON GAMMA‐1B,RECOMB ADCIRCA TADALAFIL ADEMPAS RIOCIGUAT AFINITOR EVEROLIMUS AFINITOR DISPERZ EVEROLIMUS ALECENSA ALECTINIB HCL ALUNBRIG BRIGATINIB ALYQ TADALAFIL AMBRISENTAN AMBRISENTAN AMPYRA DALFAMPRIDINE APOKYN APOMORPHINE HCL ARANESP DARBEPOETIN ALFA IN POLYSORBAT ARCALYST RILONACEPT ARIKAYCE AMIKACIN LIPOSOMAL/NEB.ACCESSR ARIXTRA FONDAPARINUX SODIUM AUBAGIO TERIFLUNOMIDE AUSTEDO DEUTETRABENAZINE AVEED TESTOSTERONE UNDECANOATE AVONEX INTERFERON BETA‐1A AVONEX INTERFERON BETA‐1A/ALBUMIN AVONEX PEN INTERFERON BETA‐1A AYVAKIT AVAPRITINIB BAFIERTAM MONOMETHYL FUMARATE BALVERSA ERDAFITINIB BENLYSTA BELIMUMAB BETASERON INTERFERON BETA‐1B BETHKIS TOBRAMYCIN BOSENTAN BOSENTAN BOSULIF BOSUTINIB BRAFTOVI ENCORAFENIB BRONCHITOL MANNITOL BRUKINSA ZANUBRUTINIB BYNFEZIA OCTREOTIDE ACETATE CABENUVA CABOTEGRAVIR/RILPIVIRINE CABLIVI CAPLACIZUMAB‐YHDP CABOMETYX CABOZANTINIB S‐MALATE CALQUENCE ACALABRUTINIB CAPECITABINE CAPECITABINE CAPRELSA VANDETANIB CARBAGLU CARGLUMIC ACID CAYSTON AZTREONAM LYSINE CERDELGA ELIGLUSTAT TARTRATE CETROTIDE CETRORELIX ACETATE CHENODAL CHENODIOL CHOLBAM CHOLIC ACID CHORIONIC GONADOTROPIN CHORIONIC GONADOTROPIN, HUMAN CIMZIA CERTOLIZUMAB PEGOL COPAXONE GLATIRAMER ACETATE COPIKTRA DUVELISIB COSENTYX (2 SYRINGES) SECUKINUMAB COSENTYX PEN SECUKINUMAB COSENTYX PEN (2 PENS) SECUKINUMAB COSENTYX SYRINGE SECUKINUMAB COTELLIC COBIMETINIB FUMARATE CYSTADANE -

Amifampridine Phosphate (Firdapse®)
AMIFAMPRIDINE PHOSPHATE (FIRDAPSEVR ) IS EFFECTIVE AND SAFE IN A PHASE 3 CLINICAL TRIAL IN LEMS SHIN J OH, MD,1 NATALYA SHCHERBAKOVA, MD,2 ANNA KOSTERA-PRUSZCZYK, MD, PhD,3 MOHAMMAD ALSHARABATI, MD,1 MAZEN DIMACHKIE, MD,4 JOSE MUNOZ BLANCO, MD,5 THOMAS BRANNAGAN, MD,6 DRAGANA LAVRNIC´ , MD, PhD,7 PERRY B SHIEH, MD, PhD,8 CHRISTOPHE VIAL, MD,9 ANDREAS MEISEL, MD,10 SAMUEL KOMOLY, MD, PhD, DSc,11 BENEDIKT SCHOSER, MD,12 KUMARASWAMY SIVAKUMAR, MD,13 YUEN SO, MD, PhD,14 and LEMS STUDY GROUP 1 Department of Neurology, University of Alabama at Birmingham, Birmingham, Alabama, USA 2 Russian Academy of Medical Sciences, Scientific Center of Neurology, Moscow, Russia 3 Department of Neurology, Medical University of Warsaw, Poland 4 University of Kansas Medical Center, Kansas City, Kansas, USA 5 Gregorio Maranon Hospital, Madrid, Spain 6 Columbia University Medical Center, New York, New York, USA 7 Clinical Center of Serbia, Clinic of Neurology, Belgrade, Serbia 8 Department of Neurology, University of California, Los Angeles, California, USA 9 Hospital of Lyon, ENMG Service and Neuromuscular Pathology Hospital, Lyon, France 10 Charite Universitatsmedizin Berlin-NeuroCure Clinical Research Center, Berlin, Germany 11 University of Pecs, Department of Neurology, Pecs, Hungary 12 Ludwig-Maximilians-University Munich Friedrich-Baur-Institute, Munich, Germany 13 Neuromuscular Research Center, Phoenix, Arizona, USA [email protected] 14 Stanford University, Stanford, California, USA Accepted 4 February 2016 ABSTRACT: Objective: We evaluated the efficacy and safety ated; the most common adverse events were oral and digital of amifampridine phosphate (FirdapseVR ) for symptomatic treat- paresthesias, nausea, and headache. Conclusions: This study ment in Lambert-Eaton myasthenic syndrome (LEMS).