Functional Analysis of Genomic Variations Associated with Emerging Artemisinin Resistant P
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Essential Function of the Alveolin Network in the Subpellicular
RESEARCH ARTICLE Essential function of the alveolin network in the subpellicular microtubules and conoid assembly in Toxoplasma gondii Nicolo` Tosetti1, Nicolas Dos Santos Pacheco1, Eloı¨se Bertiaux2, Bohumil Maco1, Lore` ne Bournonville2, Virginie Hamel2, Paul Guichard2, Dominique Soldati-Favre1* 1Department of Microbiology and Molecular Medicine, Faculty of Medicine, University of Geneva, Geneva, Switzerland; 2Department of Cell Biology, Sciences III, University of Geneva, Geneva, Switzerland Abstract The coccidian subgroup of Apicomplexa possesses an apical complex harboring a conoid, made of unique tubulin polymer fibers. This enigmatic organelle extrudes in extracellular invasive parasites and is associated to the apical polar ring (APR). The APR serves as microtubule- organizing center for the 22 subpellicular microtubules (SPMTs) that are linked to a patchwork of flattened vesicles, via an intricate network composed of alveolins. Here, we capitalize on ultrastructure expansion microscopy (U-ExM) to localize the Toxoplasma gondii Apical Cap protein 9 (AC9) and its partner AC10, identified by BioID, to the alveolin network and intercalated between the SPMTs. Parasites conditionally depleted in AC9 or AC10 replicate normally but are defective in microneme secretion and fail to invade and egress from infected cells. Electron microscopy revealed that the mature parasite mutants are conoidless, while U-ExM highlighted the disorganization of the SPMTs which likely results in the catastrophic loss of APR and conoid. Introduction *For correspondence: Toxoplasma gondii belongs to the phylum of Apicomplexa that groups numerous parasitic protozo- Dominique.Soldati-Favre@unige. ans causing severe diseases in humans and animals. As part of the superphylum of Alveolata, the ch Apicomplexa are characterized by the presence of the alveoli, which consist in small flattened single- membrane sacs, underlying the plasma membrane (PM) to form the inner membrane complex (IMC) Competing interest: See of the parasite. -

Extra-Intestinal Coccidians Plasmodium Species Distribution Of
Extra-intestinal coccidians Apicomplexa Coccidia Gregarinea Piroplasmida Eimeriida Haemosporida -Eimeriidae -Theileriidae -Haemosporiidae -Cryptosporidiidae - Babesiidae (Plasmodium) -Sarcocystidae (Sacrocystis) Aconoid (Toxoplasmsa) Plasmodium species Causitive agent of Malaria ~155 species named Infect birds, reptiles, rodents, primates, humans Species is specific for host and •P. falciparum vector •P. vivax 4 species cause human disease •P. malariae No zoonoses or animal reservoirs •P. ovale Transmission by Anopheles mosquito Distribution of Malarial Parasites P. vivax most widespread, found in most endemic areas including some temperate zones P. falciparum primarily tropics and subtropics P. malariae similar range as P. falciparum, but less common and patchy distribution P. ovale occurs primarily in tropical west Africa 1 Distribution of Malaria US Army, 1943 300 - 500 million cases per year 1.5 to 2.0 million deaths per year #1 cause of infant mortality in Africa! 40% of world’s population is at risk Malaria Atlas Map Project http://www.map.ox.ac.uk/index.htm 2 Malaria in the United States Malaria was quite prevalent in the rural South It was eradicated after world war II in an aggressive campaign using, treatment, vector control and exposure control Time magazine - 1947 (along with overall improvement of living Was a widely available, conditions) cheap insecticide This was the CDCs initial DDT resistance misssion Half-life in mammals - 8 years! US banned use of DDT in 1973 History of Malaria Considered to be the most -

A MOLECULAR PHYLOGENY of MALARIAL PARASITES RECOVERED from CYTOCHROME B GENE SEQUENCES
J. Parasitol., 88(5), 2002, pp. 972±978 q American Society of Parasitologists 2002 A MOLECULAR PHYLOGENY OF MALARIAL PARASITES RECOVERED FROM CYTOCHROME b GENE SEQUENCES Susan L. Perkins* and Jos. J. Schall Department of Biology, University of Vermont, Burlington, Vermont 05405. e-mail: [email protected] ABSTRACT: A phylogeny of haemosporidian parasites (phylum Apicomplexa, family Plasmodiidae) was recovered using mito- chondrial cytochrome b gene sequences from 52 species in 4 genera (Plasmodium, Hepatocystis, Haemoproteus, and Leucocy- tozoon), including parasite species infecting mammals, birds, and reptiles from over a wide geographic range. Leucocytozoon species emerged as an appropriate out-group for the other malarial parasites. Both parsimony and maximum-likelihood analyses produced similar phylogenetic trees. Life-history traits and parasite morphology, traditionally used as taxonomic characters, are largely phylogenetically uninformative. The Plasmodium and Hepatocystis species of mammalian hosts form 1 well-supported clade, and the Plasmodium and Haemoproteus species of birds and lizards form a second. Within this second clade, the relation- ships between taxa are more complex. Although jackknife support is weak, the Plasmodium of birds may form 1 clade and the Haemoproteus of birds another clade, but the parasites of lizards fall into several clusters, suggesting a more ancient and complex evolutionary history. The parasites currently placed within the genus Haemoproteus may not be monophyletic. Plasmodium falciparum of humans was not derived from an avian malarial ancestor and, except for its close sister species, P. reichenowi,is only distantly related to haemospordian parasites of all other mammals. Plasmodium is paraphyletic with respect to 2 other genera of malarial parasites, Haemoproteus and Hepatocystis. -

Equine Piroplasmosis
EAZWV Transmissible Disease Fact Sheet Sheet No. 119 EQUINE PIROPLASMOSIS ANIMAL TRANS- CLINICAL SIGNS FATAL TREATMENT PREVENTION GROUP MISSION DISEASE ? & CONTROL AFFECTED Equines Tick-borne Acute, subacute Sometimes Babesiosis: In houses or chronic disease fatal, in Imidocarb Tick control characterised by particular in (Imizol, erythrolysis: fever, acute T.equi Carbesia, in zoos progressive infections. Forray) Tick control anaemia, icterus, When Dimenazene haemoglobinuria haemoglobinuria diaceturate (in advanced develops, (Berenil) stages). prognosis is Theileriosis: poor. Buparvaquone (Butalex) Fact sheet compiled by Last update J. Brandt, Royal Zoological Society of Antwerp, February 2009 Belgium Fact sheet reviewed by D. Geysen, Animal Health, Institute of Tropical Medicine, Antwerp, Belgium F. Vercammen, Royal Zoological Society of Antwerp, Belgium Susceptible animal groups Horse (Equus caballus), donkey (Equus asinus), mule, zebra (Equus zebra) and Przewalski (Equus przewalskii), likely all Equus spp. are susceptible to equine piroplasmosis or biliary fever. Causative organism Babesia caballi: belonging to the phylum of the Apicomplexa, order Piroplasmida, family Babesiidae; Theileria equi, formerly known as Babesia equi or Nutallia equi, apicomplexa, order Piroplasmida, family Theileriidae. Babesia canis has been demonstrated by molecular diagnosis in apparently asymptomatic horses. A single case of Babesia bovis and two cases of Babesia bigemina have been detected in horses by a quantitative PCR. Zoonotic potential Equine piroplasmoses are specific for Equus spp. yet there are some reports of T.equi in asymptomatic dogs. Distribution Widespread: B.caballi occurs in southern Europe, Russia, Asia, Africa, South and Central America and the southern states of the US. T.equi has a more extended geographical distribution and even in tropical regions it occurs more frequent than B.caballi, also in the Mediterranean basin, Switzerland and the SW of France. -

First Case of Autochthonous Equine Theileriosis in Austria
pathogens Case Report First Case of Autochthonous Equine Theileriosis in Austria Esther Dirks 1, Phebe de Heus 1, Anja Joachim 2, Jessika-M. V. Cavalleri 1 , Ilse Schwendenwein 3, Maria Melchert 4 and Hans-Peter Fuehrer 2,* 1 Clinical Unit of Equine Internal Medicine, Department Hospital for Companion Animals and Horses, University of Veterinary Medicine Vienna, 1210 Vienna, Austria; [email protected] (E.D.); [email protected] (P.d.H.); [email protected] (J.-M.V.C.) 2 Department of Pathobiology, Institute of Parasitology, University of Veterinary Medicine Vienna, 1210 Vienna, Austria; [email protected] 3 Clinical Pathology Platform, Department of Pathobiology, University of Veterinary Medicine Vienna, 1210 Vienna, Austria; [email protected] 4 Centre for Insemination and Embryo transfer Platform, Department Hospital for Companion Animals and Horses, University of Veterinary Medicine Vienna, 1210 Vienna, Austria; [email protected] * Correspondence: [email protected]; Tel.: +43-125-077-2205 Abstract: A 23-year-old pregnant warmblood mare from Güssing, Eastern Austria, presented with apathy, anemia, fever, tachycardia and tachypnoea, and a severely elevated serum amyloid A concentration. The horse had a poor body condition and showed thoracic and pericardial effusions, and later dependent edema and icteric mucous membranes. Blood smear and molecular analyses revealed an infection with Theileria equi. Upon treatment with imidocarb diproprionate, the mare improved clinically, parasites were undetectable in blood smears, and 19 days after hospitalization the horse was discharged from hospital. However, 89 days after first hospitalization, the mare again presented to the hospital with an abortion, and the spleen of the aborted fetus was also PCR-positive Citation: Dirks, E.; de Heus, P.; for T. -

Cytauxzoon Sp.: Un Protozoo Emergente Nel Gatto
Sede amministrativa: Università degli studi di Padova Dipartimento di Biomedicina Comparata e Alimentazione Scuola di Dottorato di ricerca in SCIENZE VETERINARIE Indirizzo: SCIENZE BIOMEDICHE VETERINARIE E COMPARATE Ciclo XXV CYTAUXZOON SP.: UN PROTOZOO EMERGENTE NEL GATTO Direttore della Scuola: Ch.mo Prof. Gianfranco Gabai Coordinatore di Indirizzo: Ch.mo Prof. Gianfranco Gabai Supervisore: Ch.mo Prof. Mario Pietrobelli Correlatori: Ch. ma Prof.ssa Gioia Capelli Dr. Tommaso Furlanello Dr.ssa Laia Solano-Gallego Dottoranda: Dr.ssa Erika Carli Alla mia famiglia e a chi se ne sente parte 2 Ringraziamenti L’ultima cosa che sto scrivendo in questa tesi è la prima che voglio sia letta e cioè questa pagina di ringraziamenti. Innanzitutto voglio ringraziare l’inventore del microscopio ottico, anche se non si sa chi sia. Galileo Galilei, uno dei possibili nomi, definì il microscopio di sua costruzione “un occhialino per vedere le cose minime”. Chi come me lavora quotidiamente con questo strumento, sa quanto sia affascinante e, a volte, esaltante poter vedere “le cose minime” e può capire quanto sia stato entusiasmante il momento in cui ho visto per la prima volta Cytauxzoon nei globuli rossi di un gatto! Incomincio con il ringraziare Marco Caldin che mi ha insegnato e trasmesso la sua passione per l’Ematologia. Lo ringrazio anche perché la Clinica San Marco, da lui diretta, ha totalmente finaziato questo progetto di ricerca. Ringrazio Mario Pietrobelli che mi ha seguita e accompagnata da buon tutor in questo percorso e che ha accolto sempre con grande entusiasmo gli annunci delle mie gravidanze. Grazie anche ai miei correlatori Tommaso Furlanello e Gioia Capelli, per avermi dato suggerimenti preziosi e spunti di riflessione. -

Highly Rearranged Mitochondrial Genome in Nycteria Parasites (Haemosporidia) from Bats
Highly rearranged mitochondrial genome in Nycteria parasites (Haemosporidia) from bats Gregory Karadjiana,1,2, Alexandre Hassaninb,1, Benjamin Saintpierrec, Guy-Crispin Gembu Tungalunad, Frederic Arieye, Francisco J. Ayalaf,3, Irene Landaua, and Linda Duvala,3 aUnité Molécules de Communication et Adaptation des Microorganismes (UMR 7245), Sorbonne Universités, Muséum National d’Histoire Naturelle, CNRS, CP52, 75005 Paris, France; bInstitut de Systématique, Evolution, Biodiversité (UMR 7205), Sorbonne Universités, Muséum National d’Histoire Naturelle, CNRS, Université Pierre et Marie Curie, CP51, 75005 Paris, France; cUnité de Génétique et Génomique des Insectes Vecteurs (CNRS URA3012), Département de Parasites et Insectes Vecteurs, Institut Pasteur, 75015 Paris, France; dFaculté des Sciences, Université de Kisangani, BP 2012 Kisangani, Democratic Republic of Congo; eLaboratoire de Biologie Cellulaire Comparative des Apicomplexes, Faculté de Médicine, Université Paris Descartes, Inserm U1016, CNRS UMR 8104, Cochin Institute, 75014 Paris, France; and fDepartment of Ecology and Evolutionary Biology, University of California, Irvine, CA 92697 Contributed by Francisco J. Ayala, July 6, 2016 (sent for review March 18, 2016; reviewed by Sargis Aghayan and Georges Snounou) Haemosporidia parasites have mostly and abundantly been de- and this lack of knowledge limits the understanding of the scribed using mitochondrial genes, and in particular cytochrome evolutionary history of Haemosporidia, in particular their b (cytb). Failure to amplify the mitochondrial cytb gene of Nycteria basal diversification. parasites isolated from Nycteridae bats has been recently reported. Nycteria parasites have been primarily described, based on Bats are hosts to a diverse and profuse array of Haemosporidia traditional taxonomy, in African insectivorous bats of two fami- parasites that remain largely unstudied. -

Great Diversity of Piroplasmida in Equidae in Africa and Europe, Including Potential New Species
Veterinary Parasitology: Regional Studies and Reports 18 (2019) 100332 Contents lists available at ScienceDirect Veterinary Parasitology: Regional Studies and Reports journal homepage: www.elsevier.com/locate/vprsr Original Article Great diversity of Piroplasmida in Equidae in Africa and Europe, including T potential new species Handi Dahmanaa, Nadia Amanzougaghenea, Bernard Davousta,b, Thomas Normandb, Olivier Caretteb, Jean-Paul Demoncheauxb, Baptiste Mulotc, Bernard Fabrizyd, Pierre Scandolab, ⁎ Makhlouf Chika, Florence Fenollare, Oleg Mediannikova, a IRD, AP-HM, MEPHI, IHU-Méditerranée Infection, Aix Marseille Univ, Marseille, France b Working Group on Animal Epidemiology, French Forces Medical Service, Toulon, France c ZoologicalResarch Center, Saint-Aignan-sur-Cher, ZooParc of Beauval, France d Clinique Vétérinaire CyrnevetLupino, Bastia, France e IRD, AP-HM, SSA, VITROME, IHU-Méditerranée Infection, Aix Marseille Univ, Marseille, France ARTICLE INFO ABSTRACT Keywords: Piroplasms are Apicomplexa tick-borne parasites distributed worldwide. They are responsible for piroplasmosis Piroplasmosis (theileriosis and babesiosis) in Vertebrata and are therefore of medical and economic importance. Equidae Herein, we developed a new real time PCR assay targeting the 5.8S rRNA gene and three standard PCR assays, PCR assays targeting 18S rRNA, 28S rRNA, and cox1 genes, for the detection of piroplasmids. These assays were first op- Sub-Saharan Africa timized and screened for specificity and sensitivity. Then, they were used to study a total of 548 blood samples France and 97 ticks collected from Equidae in four sub-Saharan countries (Senegal, Democratic Republic of the Congo, Chad, and Djibouti) and France (Marseille and Corsica). DNA of piroplasms was detected in 162 of 548 (29.5%) blood samples and in 9 of 97 (9.3%) ticks. -
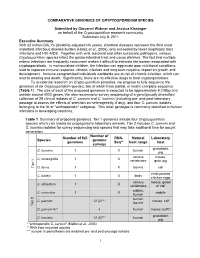
1 Comparative Genomics of Cryptosporidium Species
COMPARATIVE GENOMICS OF CRYPTOSPORIDIUM SPECIES Submitted by Giovanni Widmer and Jessica Kissinger on behalf of the Cryptosporidium research community Submitted July 8, 2011 Executive Summary With 62 million DALYs (disability-adjusted life years), diarrheal diseases represent the third most important infectious disease burden (Hotez et al., 2006), only exceeded by lower respiratory tract infections and HIV-AIDS. Together with viral, bacterial and other eukaryotic pathogens, various Cryptosporidium species infect the gastro-intestinal tract and cause diarrhea. The fact that multiple enteric infections are frequently concurrent makes it difficult to estimate the burden associated with cryptosporidiosis. In malnourished children, the infection can aggravate poor nutritional conditions, lead to impaired immune response, chronic infection and long-term negative impact on growth and development. Immune-compromised individuals worldwide are at risk of chronic infection, which can lead to wasting and death. Significantly, there are no effective drugs to treat cryptosporidiosis. To accelerate research on Cryptosporidium parasites, we propose to fully sequence the genomes of six Cryptosporidium species, two of which have partial, or nearly complete sequence (Table 1). The size of each of the proposed genomes is expected to be approximately 9.2 Mbp and contain around 4000 genes. We also recommend survey sequencing of a genotypically diversified collection of 28 clinical isolates of C. parvum and C. hominis (including pre- and post-laboratory passage to assess the effects of selection on heterogeneity, if any), and four C. parvum isolates belonging to the IIc or "anthroponotic" subgroup. This latter genotype is commonly identified in human infections in developing countries. Table 1 Summary of proposed genomes. -

Systema Naturae. the Classification of Living Organisms
Systema Naturae. The classification of living organisms. c Alexey B. Shipunov v. 5.601 (June 26, 2007) Preface Most of researches agree that kingdom-level classification of living things needs the special rules and principles. Two approaches are possible: (a) tree- based, Hennigian approach will look for main dichotomies inside so-called “Tree of Life”; and (b) space-based, Linnaean approach will look for the key differences inside “Natural System” multidimensional “cloud”. Despite of clear advantages of tree-like approach (easy to develop rules and algorithms; trees are self-explaining), in many cases the space-based approach is still prefer- able, because it let us to summarize any kinds of taxonomically related da- ta and to compare different classifications quite easily. This approach also lead us to four-kingdom classification, but with different groups: Monera, Protista, Vegetabilia and Animalia, which represent different steps of in- creased complexity of living things, from simple prokaryotic cell to compound Nature Precedings : doi:10.1038/npre.2007.241.2 Posted 16 Aug 2007 eukaryotic cell and further to tissue/organ cell systems. The classification Only recent taxa. Viruses are not included. Abbreviations: incertae sedis (i.s.); pro parte (p.p.); sensu lato (s.l.); sedis mutabilis (sed.m.); sedis possi- bilis (sed.poss.); sensu stricto (s.str.); status mutabilis (stat.m.); quotes for “environmental” groups; asterisk for paraphyletic* taxa. 1 Regnum Monera Superphylum Archebacteria Phylum 1. Archebacteria Classis 1(1). Euryarcheota 1 2(2). Nanoarchaeota 3(3). Crenarchaeota 2 Superphylum Bacteria 3 Phylum 2. Firmicutes 4 Classis 1(4). Thermotogae sed.m. 2(5). -

Genome-Wide Identification and Evolutionary Analysis of Sarcocystis Neurona Protein Kinases
Article Genome-Wide Identification and Evolutionary Analysis of Sarcocystis neurona Protein Kinases Edwin K. Murungi 1,* and Henry M. Kariithi 2 1 Department of Biochemistry and Molecular Biology, Egerton University, P.O. Box 536, 20115 Njoro, Kenya 2 Biotechnology Research Institute, Kenya Agricultural and Livestock Research Organization, P.O. Box 57811, Kaptagat Rd, Loresho, 00200 Nairobi, Kenya; [email protected] * Correspondence: [email protected]; Tel: +254-789-716-059 Academic Editor: Anthony Underwood Received: 6 January 2017; Accepted: 17 March 2017; Published: 21 March 2017 Abstract: The apicomplexan parasite Sarcocystis neurona causes equine protozoal myeloencephalitis (EPM), a degenerative neurological disease of horses. Due to its host range expansion, S. neurona is an emerging threat that requires close monitoring. In apicomplexans, protein kinases (PKs) have been implicated in a myriad of critical functions, such as host cell invasion, cell cycle progression and host immune response evasion. Here, we used various bioinformatics methods to define the kinome of S. neurona and phylogenetic relatedness of its PKs to other apicomplexans. We identified 97 putative PKs clustering within the various eukaryotic kinase groups. Although containing the universally-conserved PKA (AGC group), S. neurona kinome was devoid of PKB and PKC. Moreover, the kinome contains the six-conserved apicomplexan CDPKs (CAMK group). Several OPK atypical kinases, including ROPKs 19A, 27, 30, 33, 35 and 37 were identified. Notably, S. neurona is devoid of the virulence-associated ROPKs 5, 6, 18 and 38, as well as the Alpha and RIO kinases. Two out of the three S. neurona CK1 enzymes had high sequence similarities to Toxoplasma gondii TgCK1-α and TgCK1-β and the Plasmodium PfCK1. -

Equine Piroplasmosis Is a Tick-Borne Protozoal Disease That Affects Horses and Other Piroplasmosis Equids
Equine Importance Equine piroplasmosis is a tick-borne protozoal disease that affects horses and other Piroplasmosis equids. The consequences of infection may include asymptomatic carriage, an acute and potentially life-threatening illness, or chronic disease with vague clinical signs such as Equine Babesiosis, Equine reduced exercise tolerance. Piroplasmosis is a significant constraint to the international Theileriosis, Biliary Fever movement of equids, limiting both trade and participation in international competitions. Approximately 90% of equids worldwide are thought to live in areas where this disease is endemic, and detecting asymptomatic carriers can be difficult. In particular, the complement fixation test used for screening imported animals in the past had a Last Updated: December 2018 significant number of false negative results. Some asymptomatic carriers screened with this test may now live in areas thought to be piroplasmosis-free. Such carriers can transmit this disease to other equids through tick vectors or procedures that transmit blood, including the reuse of needles. Etiology Equine piroplasmosis can be caused by two protozoa, Babesia caballi and Theileria equi (formerly Babesia equi), both members of the phylum Apicomplexa and order Piroplasmida. T. equi is a very diverse species and has been divided into at least 3 major genotypes, A through C. Concurrent infections with B. caballi and T. equi are possible. An organism related to T. equi was described in North American horses in 2018 and proposed as a new species, Theileria haneyi. Its clinical significance is currently unclear. Species of Babesia and Theileria that are normally found in other animal hosts have been reported occasionally in equids.