The Endangered White Sands Pupfish (Cyprinodon Tularosa)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Introductions of Threatened and Endangered Fishes (Full Statement)
AFS Policy Statement #19: Introductions of Threatened and Endangered Fishes (Full Statement) ABSTRACT Introductions of threatened and endangered fishes are often an integral feature in their recovery programs. More than 80% of threatened and endangered fishes have recovery plans that call for introductions to establish a new population or an educational exhibit, supplement an existing population, or begin artificial propagation. Despite a large number of recent and proposed introductions, no systematic procedural policies have been developed to conduct these recovery efforts. Some introductions have been inadequately planned or poorly implemented. As a result, introductions of some rare fishes have been successful, whereas recovery for others has progressed slowly. In at least one instance, the introduced fish eliminated a population of another rare native organism. We present guidelines for introductions of endangered and threatened fishes that are intended to apply when an introduction is proposed to supplement an existing population or establish a new population. However, portions of the guidelines may be helpful in other Situations, such as establishing a hatchery stock. The guidelines are divided into three components: (1) selecting the introduction site, (2) conducting the introduction, and (3) post-introduction monitoring, reporting, and analysis. Implementation should increase success of efforts to recover rare fishes. "On 3 August 1968, we collected 30 or 40 individuals from among the inundated prickly pear and mesquite near the flooded spring, which by that time was covered with about 7 m of clear water." Peden (1973) The above quote described the collection of Amistad gambusia, Gambusia amistadensis, as its habitat was being flooded. Fortunately, most translocations of endangered fishes do not occur under such a feverish pace as did this collection of Amistad gambusia. -

Alien Freshwater Fish, Xiphophorus Interspecies Hybrid (Poeciliidae) Found in Artificial Lake in Warsaw, Central Poland
Available online at www.worldscientificnews.com WSN 132 (2019) 291-299 EISSN 2392-2192 SHORT COMMUNICATION Alien freshwater fish, Xiphophorus interspecies hybrid (Poeciliidae) found in artificial lake in Warsaw, Central Poland Rafał Maciaszek1,*, Dorota Marcinek2, Maria Eberhardt3, Sylwia Wilk4 1 Department of Genetics and Animal Breeding, Faculty of Animal Sciences, Warsaw University of Life Sciences, ul. Ciszewskiego 8, 02-786 Warsaw, Poland 2 Faculty of Animal Sciences, Warsaw University of Life Sciences, ul. Ciszewskiego 8, 02-786 Warsaw, Poland 3 Faculty of Veterinary Medicine, Warsaw University of Life Sciences, ul. Ciszewskiego 8, 02-786 Warsaw, Poland 4 Veterinary Clinic “Lavia-Vet”, Jasionka 926, 36-002, Jasionka, Poland *E-mail address: [email protected] ABSTRACT This paper describes an introduction of aquarium ornamental fish, Xiphophorus interspecies hybrid (Poeciliidae) in an artificial water reservoir in Pole Mokotowskie park complex in Warsaw, Poland. Caught individuals have been identified, described and presented in photographs. Measurements of selected physicochemical parameters of water were made and perspectives for the studied population were evaluated. The finding is discussed with available literature describing introductions of alien species with aquaristical origin in Polish waters. Keywords: aquarium, invasive species, ornamental pet, green swordtail, southern platyfish, variatus platy, stone maroko, Pole Mokotowskie park complex, Xiphophorus ( Received 14 July 2019; Accepted 27 July 2019; Date of Publication 29 July 2019 ) World Scientific News 132 (2019) 291-299 1. INTRODUCTION The fish kept in aquariums and home ponds are often introduced to new environment accidentaly or intentionaly by irresponsible owners. Some species of these ornamental animals are characterized by high expansiveness and tolerance to water pollution, which in the case of their release in a new area may result in local ichthyofauna biodiversity decline. -
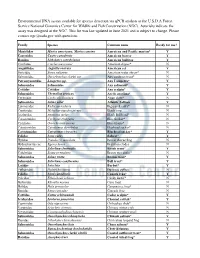
Edna Assay Development
Environmental DNA assays available for species detection via qPCR analysis at the U.S.D.A Forest Service National Genomics Center for Wildlife and Fish Conservation (NGC). Asterisks indicate the assay was designed at the NGC. This list was last updated in June 2021 and is subject to change. Please contact [email protected] with questions. Family Species Common name Ready for use? Mustelidae Martes americana, Martes caurina American and Pacific marten* Y Castoridae Castor canadensis American beaver Y Ranidae Lithobates catesbeianus American bullfrog Y Cinclidae Cinclus mexicanus American dipper* N Anguillidae Anguilla rostrata American eel Y Soricidae Sorex palustris American water shrew* N Salmonidae Oncorhynchus clarkii ssp Any cutthroat trout* N Petromyzontidae Lampetra spp. Any Lampetra* Y Salmonidae Salmonidae Any salmonid* Y Cottidae Cottidae Any sculpin* Y Salmonidae Thymallus arcticus Arctic grayling* Y Cyrenidae Corbicula fluminea Asian clam* N Salmonidae Salmo salar Atlantic Salmon Y Lymnaeidae Radix auricularia Big-eared radix* N Cyprinidae Mylopharyngodon piceus Black carp N Ictaluridae Ameiurus melas Black Bullhead* N Catostomidae Cycleptus elongatus Blue Sucker* N Cichlidae Oreochromis aureus Blue tilapia* N Catostomidae Catostomus discobolus Bluehead sucker* N Catostomidae Catostomus virescens Bluehead sucker* Y Felidae Lynx rufus Bobcat* Y Hylidae Pseudocris maculata Boreal chorus frog N Hydrocharitaceae Egeria densa Brazilian elodea N Salmonidae Salvelinus fontinalis Brook trout* Y Colubridae Boiga irregularis Brown tree snake* -

Endangered Species
FEATURE: ENDANGERED SPECIES Conservation Status of Imperiled North American Freshwater and Diadromous Fishes ABSTRACT: This is the third compilation of imperiled (i.e., endangered, threatened, vulnerable) plus extinct freshwater and diadromous fishes of North America prepared by the American Fisheries Society’s Endangered Species Committee. Since the last revision in 1989, imperilment of inland fishes has increased substantially. This list includes 700 extant taxa representing 133 genera and 36 families, a 92% increase over the 364 listed in 1989. The increase reflects the addition of distinct populations, previously non-imperiled fishes, and recently described or discovered taxa. Approximately 39% of described fish species of the continent are imperiled. There are 230 vulnerable, 190 threatened, and 280 endangered extant taxa, and 61 taxa presumed extinct or extirpated from nature. Of those that were imperiled in 1989, most (89%) are the same or worse in conservation status; only 6% have improved in status, and 5% were delisted for various reasons. Habitat degradation and nonindigenous species are the main threats to at-risk fishes, many of which are restricted to small ranges. Documenting the diversity and status of rare fishes is a critical step in identifying and implementing appropriate actions necessary for their protection and management. Howard L. Jelks, Frank McCormick, Stephen J. Walsh, Joseph S. Nelson, Noel M. Burkhead, Steven P. Platania, Salvador Contreras-Balderas, Brady A. Porter, Edmundo Díaz-Pardo, Claude B. Renaud, Dean A. Hendrickson, Juan Jacobo Schmitter-Soto, John Lyons, Eric B. Taylor, and Nicholas E. Mandrak, Melvin L. Warren, Jr. Jelks, Walsh, and Burkhead are research McCormick is a biologist with the biologists with the U.S. -

Relation of Desert Pupfish Abundance to Selected Environmental Variables
Environmental Biology of Fishes (2005) 73: 97–107 Ó Springer 2005 Relation of desert pupfish abundance to selected environmental variables in natural and manmade habitats in the Salton Sea basin Barbara A. Martin & Michael K. Saiki U.S. Geological Survey, Biological Resources Division, Western Fisheries Research Center-Dixon Duty Station, 6924 Tremont Road, Dixon, CA 95620, U.S.A. (e-mail: [email protected]) Received 6 April 2004 Accepted 12 October 2004 Key words: species assemblages, predation, water quality, habitat requirements, ecological interactions, endangered species Synopsis We assessed the relation between abundance of desert pupfish, Cyprinodon macularius, and selected biological and physicochemical variables in natural and manmade habitats within the Salton Sea Basin. Field sampling in a natural tributary, Salt Creek, and three agricultural drains captured eight species including pupfish (1.1% of the total catch), the only native species encountered. According to Bray– Curtis resemblance functions, fish species assemblages differed mostly between Salt Creek and the drains (i.e., the three drains had relatively similar species assemblages). Pupfish numbers and environmental variables varied among sites and sample periods. Canonical correlation showed that pupfish abundance was positively correlated with abundance of western mosquitofish, Gambusia affinis, and negatively correlated with abundance of porthole livebearers, Poeciliopsis gracilis, tilapias (Sarotherodon mossambica and Tilapia zillii), longjaw mudsuckers, Gillichthys mirabilis, and mollies (Poecilia latipinna and Poecilia mexicana). In addition, pupfish abundance was positively correlated with cover, pH, and salinity, and negatively correlated with sediment factor (a measure of sediment grain size) and dissolved oxygen. Pupfish abundance was generally highest in habitats where water quality extremes (especially high pH and salinity, and low dissolved oxygen) seemingly limited the occurrence of nonnative fishes. -

Cyprinodon Nevadensis Mionectes Ash Meadows Amargosa Pupfish
Ash Meadows Amargosa pupfsh Cyprinodon nevadensis mionectes WAP 2012 species due to impacts from introduced detrimental aquatc species, habitat degradaton, and federal endangered status. Agency Status NV Natural Heritage G2T2S2 USFWS LE BLM-NV Sensitve State Prot Threatened Fish NAC 503.065.3 CCVI Presumed Stable TREND: Trend is stable to increasing with contnued on-going restoraton actvites. DISTRIBUTION: Springs and associated springbrooks, outlow stream systems and terminal marshes within Ash Meadows Natonal Wildlife Refuge, Nye Co., NV. GENERAL HABITAT AND LIFE HISTORY: This species is isolated to warm springs and outlows in Ash Meadows NWR including Point of Rocks, Crystal Springs, and the Carson Slough drainage. Pupfshes feed generally on substrate; feeding territories are ofen defended by pupfshes. Diet consists of mainly algae and detritus however, aquatc insects, crustaceans, snails and eggs are also consumed. Spawning actvity is typically from February to September and in some cases year round. Males defend territories vigorously during breeding season (Soltz and Naiman 1978). In warm springs, fsh may reach sexual maturity in 4-6 weeks. Reproducton variable: in springs, pupfsh breed throughout the year, may have 8-10 generatons/year; in streams, breeds in spring and summer, 2-3 generatons/year (Moyle 1976). In springs, males establish territories over sites suitable for ovipositon. Short generaton tme allows small populatons to be viable. Young adults typically comprise most of the biomass of a populaton. Compared to other C. nevadensis subspecies, this pupfsh has a short deep body and long head with typically low fn ray and scale counts (Soltz and Naiman 1978). CONSERVATION CHALLENGES: Being previously threatened by agricultural use of the area (loss and degradaton of habitat resultng from water diversion and pumping) and by impending residental development, the TNC purchased property, which later became the Ash Meadows NWR. -

Recovery Plan Endangered and Species Nevada
RECOVERYPLAN FOR THE ENDANGEREDAND THREATENED SPECIES OF&H MEADOWS, NEVADA Prepared by Don W. Sada U.S. Fish and Wildlife Service Reno, Nevada RECOVERY PLAN FOR THE ENDANGERED AND THREATENED SPECIES OF ASH MEADOWS, NEVADA Prepared By Don W. Sada U.S. Fish and Wildlife Service Reno, Nevada for the U.S. Fish and Wildlife Service Portland, Oregon ~FP2 3 ‘:XN Date This plan covers the following federally listed species in Ash Meadows, Nevada and California: Devil’s Hole pupfish, Warm Springs pupfish, Ash Meadows Arnargosa pupfish, Ash Meadows speckled dace, Ash Meadows naucorid, Ash Meadows blazing star, Ash Meadows ivesia, Ainargosa niterwort, Spring-loving centaury, Ash Meadows sunray, Ash Meadows inilk-vetch, and Ash Meadows guxnplant. THIS IS THE COMPLETED ASH MEADOWS SPECIES RECOVERY PLAN. IT HAS BEEN APPROVED BY THE U.S. FISH AND WILDLIFE SERVICE. IT DOES NOT NECESSARILY REPRESENT OFFICIAL POSITIONS OR APPROVALS OF COOPERATING AGENCIES (AND IT DOES NOT NECESSARILY REPRESENT THE VIEWS OF ALL INDIVIDUALS) WHO PLAYED THE KEY ROLE IN PREPARING THIS PLAN. THIS PLAN IS SUBJECT TO MODIFICATION AS DICTATED BY NEW FINDINGS AND CHANGES IN SPECIES STATUS, AND COMPLETION OF TASKS DESCRIBED IN THE PLAN. GOALS AND OBJECTIVES WILL BE ATTAINED AND FUNDS EXPENDED CONTINGENT UPON APPROPRIATIONS, PRIORITIES, AND OTHER BUDGETARY CONSTRAINTS. LITERATURE CITATION SHOULD READ AS FOLLOWS U.S. Fish and Wildlife Service. 1990. Recovery plan for the endangered and threatened species of Ash Meadows, Nevada. U.S. Fish and Wildlife Service, Portland, Oregon. 123 pp. Additional copies may be obtained from Fish and Wildlife Reference Service 5430 Grosvenor Lane, Suite 110 Bethesda, Maryland 20814 Telephone: 301-492-6403 1-800-582-3421 : ACKNOWLEDGMENTS: This plan results from the efforts of many who spent considerable time and energy to prevent the destruction of Ash Meadows and the extinction of its diverse endemic biota. -

Three New Pupfish Species, Cyprinodon (Teleostei, Cyprinodontidae), from Chihuahua, México, and Arizona, USA Author(S): W
Three New Pupfish Species, Cyprinodon (Teleostei, Cyprinodontidae), from Chihuahua, México, and Arizona, USA Author(s): W. L. Minckley, Robert Rush Miller and Steven Mark Norris Source: Copeia, Vol. 2002, No. 3 (Aug. 15, 2002), pp. 687-705 Published by: American Society of Ichthyologists and Herpetologists (ASIH) Stable URL: http://www.jstor.org/stable/1448150 . Accessed: 23/07/2014 15:57 Your use of the JSTOR archive indicates your acceptance of the Terms & Conditions of Use, available at . http://www.jstor.org/page/info/about/policies/terms.jsp . JSTOR is a not-for-profit service that helps scholars, researchers, and students discover, use, and build upon a wide range of content in a trusted digital archive. We use information technology and tools to increase productivity and facilitate new forms of scholarship. For more information about JSTOR, please contact [email protected]. American Society of Ichthyologists and Herpetologists (ASIH) is collaborating with JSTOR to digitize, preserve and extend access to Copeia. http://www.jstor.org This content downloaded from 128.123.44.23 on Wed, 23 Jul 2014 15:57:21 PM All use subject to JSTOR Terms and Conditions Copeia,2002(3), pp. 687-705 Three New Pupfish Species, Cyprinodon(Teleostei, Cyprinodontidae), from Chihuahua, Mexico, and Arizona, USA W. L. MINCKLEY,ROBERT RUSH MILLER,AND STEVENMARK NORRIS Three new species of Cyprinodon(Teleostei, Cyprinodontidae)are described,each long recognized as distinct. Cyprinodonpisteri occupies a varietyof systems and hab- itats in the Lago de Guzmin complex basin in northern Chihuahua,Mexico. It is distinguishedby its dusky to black dorsal fin and narrowor inconspicuousterminal bar on the caudal fin in mature males. -

Summary Report of Freshwater Nonindigenous Aquatic Species in U.S
Summary Report of Freshwater Nonindigenous Aquatic Species in U.S. Fish and Wildlife Service Region 4—An Update April 2013 Prepared by: Pam L. Fuller, Amy J. Benson, and Matthew J. Cannister U.S. Geological Survey Southeast Ecological Science Center Gainesville, Florida Prepared for: U.S. Fish and Wildlife Service Southeast Region Atlanta, Georgia Cover Photos: Silver Carp, Hypophthalmichthys molitrix – Auburn University Giant Applesnail, Pomacea maculata – David Knott Straightedge Crayfish, Procambarus hayi – U.S. Forest Service i Table of Contents Table of Contents ...................................................................................................................................... ii List of Figures ............................................................................................................................................ v List of Tables ............................................................................................................................................ vi INTRODUCTION ............................................................................................................................................. 1 Overview of Region 4 Introductions Since 2000 ....................................................................................... 1 Format of Species Accounts ...................................................................................................................... 2 Explanation of Maps ................................................................................................................................ -

Distribution of Amargosa River Pupfish (Cyprinodon Nevadensis Amargosae) in Death Valley National Park, CA
California Fish and Game 103(3): 91-95; 2017 Distribution of Amargosa River pupfish (Cyprinodon nevadensis amargosae) in Death Valley National Park, CA KRISTEN G. HUMPHREY, JAMIE B. LEAVITT, WESLEY J. GOLDSMITH, BRIAN R. KESNER, AND PAUL C. MARSH* Native Fish Lab at Marsh & Associates, LLC, 5016 South Ash Avenue, Suite 108, Tempe, AZ 85282, USA (KGH, JBL, WJG, BRK, PCM). *correspondent: [email protected] Key words: Amargosa River pupfish, Death Valley National Park, distribution, endangered species, monitoring, intermittent streams, range ________________________________________________________________________ Amargosa River pupfish (Cyprinodon nevadensis amargosae), is one of six rec- ognized subspecies of Amargosa pupfish (Miller 1948) and survives in waters embedded in a uniquely harsh environment, the arid and hot Mojave Desert (Jaeger 1957). All are endemic to the Amargosa River basin of southern California and Nevada (Moyle 2002). Differing from other spring-dwelling subspecies of Amargosa pupfish (Cyprinodon ne- vadensis), Amargosa River pupfish is riverine and the most widely distributed, the extent of which has been underrepresented prior to this study (Moyle et al. 2015). Originating on Pahute Mesa, Nye County, Nevada, the Amargosa River flows intermittently, often under- ground, south past the towns of Beatty, Shoshone, and Tecopa and through the Amargosa River Canyon before turning north into Death Valley National Park and terminating at Badwater Basin (Figure 1). Amargosa River pupfish is data deficient with a distribution range that is largely unknown. The species has been documented in Tecopa Bore near Tecopa, Inyo County, CA (Naiman 1976) and in the Amargosa River Canyon, Inyo and San Bernardino Counties, CA (Williams-Deacon et al. -

The Extinction of the Catarina Pupfish Megupsilon Aporus and the Implications for the Conservation of Freshwater Fish in Mexico
The extinction of the Catarina pupfish Megupsilon aporus and the implications for the conservation of freshwater fish in Mexico A RCADIO V ALDÉS G ONZÁLEZ,LOURDES M ARTÍNEZ E STÉVEZ M A .ELENA Á NGELES V ILLEDA and G ERARDO C EBALLOS Abstract Extinctions are occurring at an unprecedented ; Régnier et al., ). Since the start of the st century it rate as a consequence of human activities. Vertebrates con- has become clear that population depletion and extinction stitute the best-known group of animals, and thus the group of both freshwater and marine fishes is a severe and wide- for which there are more accurate estimates of extinctions. spread problem (e.g. Ricciardi & Rasmussen, ; Myers Among them, freshwater fishes are particularly threatened &Worm,; Olden et al., ; Burkhead, ). and many species are declining. Here we report the extinc- Extinction of freshwater fishes has been relatively well tion of an endemic freshwater fish of Mexico, the Catarina documented in North America (e.g. Miller et al., ; pupfish Megupsilon aporus, the sole species of the genus Burkhead, ). A compilation of the conservation status Megupsilon. We present a synopsis of the discovery and de- of freshwater fishes in Mexico has revealed that species scription of the species, the threats to, and degradation of, its have become extinct in the wild or have been extirpated habitat, and the efforts to maintain the species in captivity from the country, and . (% of all species in before it became extinct in . The loss of the Catarina Mexico) are facing extinction (IUCN, ; Ceballos et al., pupfish has evolutionary and ecological implications, and b; Table ). -

The Effects of Introduced Tilapias on Native Biodiversity
AQUATIC CONSERVATION: MARINE AND FRESHWATER ECOSYSTEMS Aquatic Conserv: Mar. Freshw. Ecosyst. 15: 463–483 (2005) Published online in Wiley InterScience (www.interscience.wiley.com). DOI: 10.1002/aqc.699 The effects of introduced tilapias on native biodiversity GABRIELLE C. CANONICOa,*, ANGELA ARTHINGTONb, JEFFREY K. MCCRARYc,d and MICHELE L. THIEMEe a Sustainable Development and Conservation Biology Program, University of Maryland, College Park, Maryland, USA b Centre for Riverine Landscapes, Faculty of Environmental Sciences, Griffith University, Australia c University of Central America, Managua, Nicaragua d Conservation Management Institute, College of Natural Resources, Virginia Tech, Blacksburg, Virginia, USA e Conservation Science Program, World Wildlife Fund, Washington, DC, USA ABSTRACT 1. The common name ‘tilapia’ refers to a group of tropical freshwater fish in the family Cichlidae (Oreochromis, Tilapia, and Sarotherodon spp.) that are indigenous to Africa and the southwestern Middle East. Since the 1930s, tilapias have been intentionally dispersed worldwide for the biological control of aquatic weeds and insects, as baitfish for certain capture fisheries, for aquaria, and as a food fish. They have most recently been promoted as an important source of protein that could provide food security for developing countries without the environmental problems associated with terrestrial agriculture. In addition, market demand for tilapia in developed countries such as the United States is growing rapidly. 2. Tilapias are well-suited to aquaculture because they are highly prolific and tolerant to a range of environmental conditions. They have come to be known as the ‘aquatic chicken’ because of their potential as an affordable, high-yield source of protein that can be easily raised in a range of environments } from subsistence or ‘backyard’ units to intensive fish hatcheries.