Cap 0 RESUMEN, OBJETIVOS Y PLAN DE TRABAJO
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Artificial Food Colours and Children Why We Want to Limit and Label Foods Containing the ‘Southampton Six’ Food Colours on the UK Market Post-Brexit
Artificial food colours and children Why we want to limit and label foods containing the ‘Southampton Six’ food colours on the UK market post-Brexit November 2020 FIRST STEPS NUTRITIONArtificial food coloursTRUST and children: page Artificial food colours and children: Why we want to limit and label foods containing the‘Southampton Six’ food colours on the UK market post-Brexit November 2020 Published by First Steps Nutrition Trust. A PDF of this resource is available on the First Steps Nutrition Trust website. www.firststepsnutrition.org The text of this resource, can be reproduced in other materials provided that the materials promote public health and make no profit, and an acknowledgement is made to First Steps Nutrition Trust. This resource is provided for information only and individual advice on diet and health should always be sought from appropriate health professionals. First Steps Nutrition Trust Studio 3.04 The Food Exchange New Covent Garden Market London SW8 5EL Registered charity number: 1146408 First Steps Nutrition Trust is a charity which provides evidence-based and independent information and support for good nutrition from pre-conception to five years of age. For more information, see our website: www.firststepsnutrition.org Acknowledgements This report was written by Rachael Wall and Dr Helen Crawley. We would like to thank Annie Seeley, Sarah Weston, Erik Millstone and Anna Rosier for their help and support with this report. Artificial food colours and children: page 1 Contents Page Executive summary 3 Recommendations -

Regulatory Information Sheet
Regulatory Information Sheet Approved Drug Colourants Listed by the European Union Colour Index Colour E Number Alternate Names Number Allura Red AC (a) E129 16035 FD&C Red #40 Aluminum*** E173 77000 -- Amaranth*** (a) E123 16185 Delisted FD&C Red #2 Annatto*** E160b 75120 Bixin, norbixin Anthocyanins (a) E163 -- -- Beetroot Red E162 -- Betanin Beta APO-8´-Carotenal E160e 40820 -- Brilliant Black BN (a) E151 28440 Black BN Brilliant Blue FCF (a) E133 42090 FD&C Blue #1 Brown HT (a) E155 20285 -- Calcium Carbonate E170 77220 -- Canthaxanthin* E161g 40850 -- Caramel,-Plain E150a -- -- Caramel,-Caustic Sulphite E150b -- -- Caramel,-Ammonia E150c -- -- Caramel, Sulphite Ammonia E150d -- -- Carmine (a) E120 75470 Carminic Acid, Cochineal Carmoisine (a) E122 14720 Azorubine Carotenes E160a 40800 / 75130 -- Chlorophylls/Chlorophyllins E140 75810 / 75815 -- Copper Complexes of E141 75815 -- Chlorophylls/Chlorophyllins(a) Curcumin (a) E100 75300 Turmeric Erythrosine*** (a) E127 45430 FD&C Red #3 Gold*** E175 77480 -- Green S (a) E142 44090 Acid Brilliant Green BS Indigotine (a) E132 73015 FD&C Blue #2, Indigo Carmine 77491 / 77492 / Iron Oxides & Hydroxides E172 Iron Oxide Red, Yellow, Black 77499 Litholrubine BK*** (a) E180 -- -- Lutein E161b -- -- Lycopene*** E160d 75125 -- Paprika Extract E160c -- Capsanthin, Capsorubin Patent Blue V (a) E131 42051 Acid Blue 3 Ponceau 4R (a) E124 16255 Cochineal Red A Page 1 of 2 Document Reference No.: GLO-10107, revision 2 Effective Date: September 2014 Reviewed Date: November 2017 This document is valid at the time of distribution. Distributed 24-Sep-2021 (UTC) E Colour Index Colour Alternate Names Number Number Quinoline Yellow** (a) E104 47005 China Yellow Riboflavins (a) E101 -- -- Silver*** E174 -- -- Sunset Yellow FCF (a) E110 15985 FD&C Yellow #6, Orange Yellow S Tartrazine (a) E102 19140 FD&C Yellow #5 Titanium Dioxide E171 77891 -- Vegetable Carbon E153 77268:1 Carbo Medicinalis Vegetalis The above list is derived from Part B, List of All Additives, from Annex II to Regulation (EC) No 1333/2008 on food additives. -

Role of Microorganisms in Biodegradation of Food Additive Azo Dyes: a Review
Vol. 19(11), pp.799-805, November, 2020 DOI: 10.5897/AJB2020.17250 Article Number: F63AA1865367 ISSN: 1684-5315 Copyright ©2020 Author(s) retain the copyright of this article African Journal of Biotechnology http://www.academicjournals.org/AJB Review Role of microorganisms in biodegradation of food additive Azo dyes: A review Fatimah Alshehrei Department of Biology, Jamum College University, Umm AlQura University, Makkah24382, Saudi Arabia. Received 22 September, 2020; Accepted 27 October, 2020 Food additives Azo dyes are synthetic compounds added to foods to impart color and improve their properties. Some azo dyes have been banned as food additives due to toxic, mutagenic, and carcinogenic side effects. Long exposure to foods containing azo dye leads to chronic toxicity. Some microorganisms are capable to degrade these dyes and convert them to aromatic amines. In human body, microbiota can play a vital role in biodegradation of azo dyes by producing azo reductase. Aromatic amines are toxic, water-soluble and well absorbed via human intestine. In the current study, the role of microorganisms in biodegradation of six dyes related to azo group was discussed. These dyes are: Tartrazine E102, Sunset Yellow E110, Ponceau E124, Azorubine E122, Amaranth E123, and Allura Red E129 which are classified as the most harmful food additive dyes. Key word: Food additive, azo dyes, microorganisms, azo reductase, aromatic amines. INTRODUCTION Food additives are synthetic compounds added to food In the USA and European countries, some azo dyes have for many proposes such as maintaining the product from been banned as food additives due to toxic, mutagenic, deterioration or improving its safety, freshness, taste, and carcinogenic side effects (Chung, 2000). -

Electro-Oxidation–Plasma Treatment for Azo Dye Carmoisine (Acid Red 14) in an Aqueous Solution
materials Article Electro-Oxidation–Plasma Treatment for Azo Dye Carmoisine (Acid Red 14) in an Aqueous Solution Héctor Barrera 1, Julián Cruz-Olivares 2, Bernardo A. Frontana-Uribe 1,3, Aarón Gómez-Díaz 4 , Pedro G. Reyes-Romero 4,* and Carlos E. Barrera-Diaz 1,2,* 1 Centro Conjunto de Investigación de Química Sustentable CCIQS, UAEM-UNAM, Carretera Toluca Atlacomulco, km 14.5, C.P. Toluca 50200, Estado de México, Mexico; [email protected] (H.B.); [email protected] (B.A.F.-U.) 2 Facultad de Química, Universidad Autónoma del Estado de México, Paseo Colón intersección Paseo Tollocan S/N, Toluca 50120, Estado de México, Mexico; [email protected] 3 Instituto de Química, Universidad Nacional Autónoma de México, Circuito Exterior, Ciudad Universitaria, Ciudad de México 04510, Mexico 4 Facultad de Ciencias, Universidad Autónoma del Estado de México, Campus El Cerrillo, Carretera Toluca - Ixtlahuaca Km 15.5, Piedras Blancas, Toluca 50200, Estado de México, Mexico; [email protected] * Correspondence: [email protected] (P.G.R.-R.); [email protected] (C.E.B.-D.) Received: 30 October 2019; Accepted: 30 December 2019; Published: 23 March 2020 Abstract: Currently, azo dye Carmoisine is an additive that is widely used in the food processing industry sector. However, limited biodegradability in the environment has become a major concern regarding the removal of azo dye. In this study, the degradation of azo dye Carmoisine (acid red 14) in an aqueous solution was studied by using a sequenced process of electro-oxidation–plasma at atmospheric pressure (EO–PAP). Both the efficiency and effectiveness of the process were compared individually. -

Biological Stains & Dyes
BIOLOGICAL STAINS & DYES Developed for Biology, microbiology & industrial applications ACRIFLAVIN ALCIAN BLUE 8GX ACRIDINE ORANGE ALIZARINE CYANINE GREEN ANILINE BLUE (SPIRIT SOLUBLE) www.lobachemie.com BIOLOGICAL STAINS & DYES Staining is an important technique used in microscopy to enhance contrast in the microscopic image. Stains and dyes are frequently used in biology and medicine to highlight structures in biological tissues. Loba Chemie offers comprehensive range of Stains and dyes, which are frequently used in Microbiology, Hematology, Histology, Cytology, Protein and DNA Staining after Electrophoresis and Fluorescence Microscopy etc. Many of our stains and dyes have specifications complying certified grade of Biological Stain Commission, and suitable for biological research. Stringent testing on all batches is performed to ensure consistency and satisfy necessary specification particularly in challenging work such as histology and molecular biology. Stains and dyes offer by Loba chemie includes Dry – powder form Stains and dyes as well as wet - ready to use solutions. Features: • Ideally suited to molecular biology or microbiology applications • Available in a wide range of innovative chemical packaging options. Range of Biological Stains & Dyes Product Code Product Name C.I. No CAS No 00590 ACRIDINE ORANGE 46005 10127-02-3 00600 ACRIFLAVIN 46000 8063-24-9 00830 ALCIAN BLUE 8GX 74240 33864-99-2 00840 ALIZARINE AR 58000 72-48-0 00852 ALIZARINE CYANINE GREEN 61570 4403-90-1 00980 AMARANTH 16185 915-67-3 01010 AMIDO BLACK 10B 20470 -

Organic Colouring Agents in the Pharmaceutical Industry
DOI: 10.1515/fv-2017-0025 FOLIA VETERINARIA, 61, 3: 32—46, 2017 ORGANIC COLOURING AGENTS IN THE PHARMACEUTICAL INDUSTRY Šuleková, M.1, Smrčová, M.1, Hudák, A.1 Heželová, M.2, Fedorová, M.3 1Department of Chemistry, Biochemistry and Biophysics, Institute of Pharmaceutical Chemistry University of Veterinary Medicine and Pharmacy, Komenského 73, 041 81 Košice 2Faculty of Metallurgy, Institute of Recycling Technologies Technical University in Košice, Letná 9, 042 00 Košice 3Department of Pharmacy and Social Pharmacy University of Veterinary Medicine and Pharmacy, Komenského 73, 041 81 Košice Slovakia [email protected] ABSTRACT INTRODUCTION Food dyes are largely used in the process of manufac- In addition to the active ingredients, various additives turing pharmaceutical products. The aim of such a pro- are used in the manufacture of pharmaceuticals. This group cedure is not only to increase the attractiveness of prod- of compounds includes dyes. A colour additive is any dye, ucts, but also to help patients distinguish between phar- pigment, or other substance that imparts colour to food, maceuticals. Various dyes, especially organic colouring drink or any non-food applications including pharma- agents, may in some cases have a negative impact on the ceuticals. Moreover, a colour additive is also any chemical human body. They are incorporated into pharmaceuti- compound that reacts with another substance and causes cal products including tablets, hard gelatine capsules or the formation of a colour [22, 56]. The pharmaceutical in- soft gelatine capsules, lozenges, syrups, etc. This article dustry employs various inorganic and, especially, organic provides an overview of the most widely used colouring dyes for this purpose. -

Analysis of Artificial Colorants in Various Food Samples Using Monolithic Silica Columns and LC-MS by Stephan Altmaier, Merck Millipore, Frankfurter Str
31 Analysis of Artificial Colorants in Various Food Samples using Monolithic Silica Columns and LC-MS by Stephan Altmaier, Merck Millipore, Frankfurter Str. 250, 64293 Darmstadt, Germany This work describes a simple and sensitive high performance liquid chromatography method with UV or mass spectrometry detection for the analysis of artificial colorants from dye classes such as azo or chinophthalone in various food samples. After a short sample preparation procedure all samples were separated on C18 reversed phase monolithic silica columns via a gradient elution profile and directly transferred to UV or MS for the analysis of the main components. This setup enabled the identification of dyes in real life samples such as beverages or sweets within very short analysis times and with a minimised sample preparation step. In the nineteenth century chemicals such as azo compounds (see Tables 1 and 2 and by an organism very easily. mercury sulphide, lead oxide, copper salts or Figure 1). They are utilised as a single Most of the current artificial colorants can now fuchsine were utilised to artificially colour colouring ingredient or as a mixture with be replaced by natural dyes very easily. food such as cheese, confectionary, pickles other colorants in a wide variety of foods and Nevertheless, for economic reasons they are [1] or wine [2]. In the end of that century the beverages. All listed dyes are nontoxic and still used to improve the attractiveness of discovery of many synthetic organic food water soluble and can therefore be excreted sweets or soft drinks towards children or of colorants allowed for more brilliant colours than traditional natural dyes. -

Domestic and Import Food Additives and Color Additives
FOOD AND DRUG ADMINISTRATION COMPLIANCE PROGRAM GUIDANCE MANUAL PROGRAM 7309.006 CHAPTER 09 – FOOD AND COLOR ADDITIVES SUBJECT: IMPLEMENTATION DATE: DOMESTIC AND IMPORT FOOD ADDITIVES AND 10/28/2019 COLOR ADDITIVES DATA REPORTING PRODUCT CODES PRODUCT/ASSIGNMENT CODES All Food Codes (except Industry 16 09006C Color Additives (seafood)) and Industry 45-46 (Food Additives) 09006F Food Additives All Food Codes (except Industry 16 (seafood)) and Industry 50 (Color Additives) FIELD REPORTING REQUIREMENTS: Report all sample collections and analytical results into the Field Accomplishment and Compliance Tracking System (FACTS). Report all inspections into eNSpect. Scan product labeling and any product brochures into eNSpect as an exhibit. Date of Issuance: 10/28/2019 Page 1 of 2 PROGRAM 7309.006 Contents PART I - BACKGROUND .................................................................................................................... 3 PART II - IMPLEMENTATION............................................................................................................ 6 Objectives .................................................................................................................................... 6 Program Management Instructions .............................................................................................. 6 PART III - INSPECTIONAL ................................................................................................................. 9 Operations ................................................................................................................................... -

The Color of Tissue Diagnostics Routine Stains, Special Stains and Ancillary Reagents
The Color of Tissue Diagnostics Routine Stains, Special Stains and Ancillary Reagents The life science business of Merck KGaA, Darmstadt, Germany operates as MilliporeSigma in the U.S. and Canada. For over years, 100routine stains, special stains and ancillary reagents have been part of the MilliporeSigma product range. This tradition and experience has made MilliporeSigma one of the world’s leading suppliers of microscopy products. The products for microscopy, a comprehensive range for classical hematology, histology, cytology, and microbiology, are constantly being expanded and adapted to the needs of the user and to comply with all relevant global regulations. Many of MilliporeSigma’s microscopy products are classified as in vitro diagnostic (IVD) medical devices. Quality Means Trust As a result of MilliporeSigma’s focus on quality control, microscopy products are renowned for excellent reproducibility of results. MilliporeSigma products are manufactured in accordance with a quality management system using raw materials and solvents that meet the most stringent quality criteria. Prior to releasing the products for particular applications, relevant chemical and physical parameters are checked along with product functionality. The methods used for testing comply with international standards. For over Contents Ancillary Reagents Microbiology 3-4 Fixing Media 28-29 Staining Solutions and Kits years, 5-6 Embedding Media 30 Staining of Mycobacteria 100 6 Decalcifiers and Tissue Softeners 30 Control Slides 7 Mounting Media Cytology 8 Immersion -

Development and Validation of a Simple and Fast Method For
IOSR Journal of Environmental Science, Toxicology and Food Technology (IOSR-JESTFT) e-ISSN: 2319-2402,p- ISSN: 2319-2399.Volume 10, Issue 9 Ver. I (Sep. 2016), PP 17-22 www.iosrjournals.org Development and validation of a simple and fast method for simultaneous determination of sunset yellow and azorubine in carbonated orange flavor soft drinks samples by high- performance liquid chromatography Mohammad Faraji1, Gholamrezza Ghasempour1, Banafshe Nasiri Sahneh1, Rika Javanshir1, Mina Alavi 1Faculty of Food Industry and Agriculture, Department of Food science & Technology, Standard Research Institute (SRI), Karaj P.O. Box 31745-139, Iran Abstract: An efficient method was developed for the simultaneous determination of sunset yellow and azorubine in carbonated orange flavor soft drink samples by high performance liquid chromatography (HPLC- UV-Vis). Affecting parameters on separation and detection of the dyes were investigated and optimized. For these dyes good linearity (0.25–50 mg L-1, > r2=0.99) were obtained. Limits of detection for both of the dyes were 0.1 mg L-1. The recoveries of the dyes ranged from 92 to 102%. Intra and inter-day precision expressed as relative standard deviation (RSD%) at 5.0 and 50 mg L-1 levels less than 8.0% were also achieved. This method has been applied successfully in the determination of the sunset yellow and azorubine in soft drink samples. The average sunset yellow concentration of the 42 soft drink samples range between 20.6- 60.2 mg L-1. Also, azorubine was found in the soft drink samples in the range of N.D-4.4 mg L-1. -
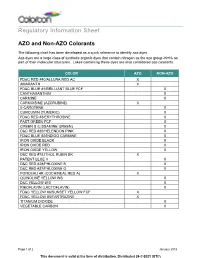
Regulatory Information Sheet AZO and Non-AZO Colorants
Regulatory Information Sheet AZO and Non-AZO Colorants The following chart has been developed as a quick reference to identify azo dyes. Azo dyes are a large class of synthetic organic dyes that contain nitrogen as the azo group -N=N- as part of their molecular structures. Lakes containing these dyes are also considered azo colorants. COLOR AZO NON-AZO FD&C RED #40/ALLURA RED AC X AMARANTH X FD&C BLUE #1/BRILLIANT BLUE FCF X CANTHAXANTHIN X CARMINE X CARMOISINE (AZORUBINE) X ß-CAROTENE X CURCUMIN (TUMERIC) X FD&C RED #3/ERYTHROSINE X FAST GREEN FCF X GREEN S (LISSAMINE GREEN) X D&C RED #30/HELENDON PINK X FD&C BLUE #2/INDIGO CARMINE X IRON OXIDE BLACK X IRON OXIDE RED X IRON OXIDE YELLOW X D&C RED #7/LITHOL RUBIN BK X PATENT BLUE V X D&C RED #28/PHLOXINE B X D&C RED #27/PHLOXINE O X PONCEAU 4R (COCHINEAL RED A) X QUINOLINE YELLOW WS X D&C YELLOW #10 X RIBOFLAVIN (LACTOFLAVIN) X FD&C YELLOW #6/SUNSET YELLOW FCF X FD&C YELLOW #5/TARTRAZINE X TITANIUM DIOXIDE X VEGETABLE CARBON X Page 1 of 2 January 2018 This document is valid at the time of distribution. Distributed 24-?-2021 (UTC) The information contained herein, to the best of our knowledge is true and accurate. Any recommendations or suggestions are made without warranty or guarantee, since the conditions of use are beyond our control. Any information contained herein is intended as a recommendation for use of our products so as not to infringe on any patent. -
Redalyc.Action of Ponceau 4R (E-124) Food Dye on Root Meristematic Cells of Allium Cepa L
Acta Scientiarum. Health Sciences ISSN: 1679-9291 [email protected] Universidade Estadual de Maringá Brasil Santana Marques, Gleuvânia; do Anjos Sousa, Josefa Janaína; Peron, Ana Paula Action of Ponceau 4R (E-124) food dye on root meristematic cells of Allium cepa L. Acta Scientiarum. Health Sciences, vol. 37, núm. 1, enero-marzo, 2015, pp. 101-106 Universidade Estadual de Maringá Maringá, Brasil Available in: http://www.redalyc.org/articulo.oa?id=307239651012 How to cite Complete issue Scientific Information System More information about this article Network of Scientific Journals from Latin America, the Caribbean, Spain and Portugal Journal's homepage in redalyc.org Non-profit academic project, developed under the open access initiative Acta Scientiarum http://www.uem.br/acta ISSN printed: 1679-9283 ISSN on-line: 1807-863X Doi: 10.4025/actascibiolsci.v37i1.23119 Action of Ponceau 4R (E-124) food dye on root meristematic cells of Allium cepa L. Gleuvânia Santana Marques, Josefa Janaína do Anjos Sousa and Ana Paula Peron* Campus Senador Helvídio Nunes de Barros, Universidade Federal do Piauí, Rua Cícero Eduardo, s/n, 64600-000, Picos, Piauí, Brazil. *Author for correspondence: E-mail: [email protected] ABSTRACT.This study aimed to evaluate the toxicity of Ponceau 4R food dye on the cell cycle in root meristematic cells of Allium cepa L. at three concentrations: 0.25, 0.50 and 0.75 g L-1, at exposure times of 24 and 48 hours. For each concentration, we used a set of five onion bulbs that were first rooted in distilled water and then transferred to their respective concentrations.