Patent Application Publication Oo) Pub. No.: US 2014/0099254 a L Chang Et Al
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Predictive QSAR Tools to Aid in Early Process Development of Monoclonal Antibodies
Predictive QSAR tools to aid in early process development of monoclonal antibodies John Micael Andreas Karlberg Published work submitted to Newcastle University for the degree of Doctor of Philosophy in the School of Engineering November 2019 Abstract Monoclonal antibodies (mAbs) have become one of the fastest growing markets for diagnostic and therapeutic treatments over the last 30 years with a global sales revenue around $89 billion reported in 2017. A popular framework widely used in pharmaceutical industries for designing manufacturing processes for mAbs is Quality by Design (QbD) due to providing a structured and systematic approach in investigation and screening process parameters that might influence the product quality. However, due to the large number of product quality attributes (CQAs) and process parameters that exist in an mAb process platform, extensive investigation is needed to characterise their impact on the product quality which makes the process development costly and time consuming. There is thus an urgent need for methods and tools that can be used for early risk-based selection of critical product properties and process factors to reduce the number of potential factors that have to be investigated, thereby aiding in speeding up the process development and reduce costs. In this study, a framework for predictive model development based on Quantitative Structure- Activity Relationship (QSAR) modelling was developed to link structural features and properties of mAbs to Hydrophobic Interaction Chromatography (HIC) retention times and expressed mAb yield from HEK cells. Model development was based on a structured approach for incremental model refinement and evaluation that aided in increasing model performance until becoming acceptable in accordance to the OECD guidelines for QSAR models. -
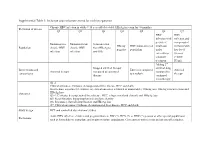
Inclusion and Exclusion Criteria for Each Key Question
Supplemental Table 1: Inclusion and exclusion criteria for each key question Chronic HBV infection in adults ≥ 18 year old (detectable HBsAg in serum for >6 months) Definition of disease Q1 Q2 Q3 Q4 Q5 Q6 Q7 HBV HBV infection with infection and persistent compensated Immunoactive Immunotolerant Seroconverted HBeAg HBV mono-infected viral load cirrhosis with Population chronic HBV chronic HBV from HBeAg to negative population under low level infection infection anti-HBe entecavir or viremia tenofovir (<2000 treatment IU/ml) Adding 2nd Stopped antiviral therapy antiviral drug Interventions and Entecavir compared Antiviral Antiviral therapy compared to continued compared to comparisons to tenofovir therapy therapy continued monotherapy Q1-2: Clinical outcomes: Cirrhosis, decompensated liver disease, HCC and death Intermediate outcomes (if evidence on clinical outcomes is limited or unavailable): HBsAg loss, HBeAg seroconversion and Outcomes HBeAg loss Q3-4: Cirrhosis, decompensated liver disease, HCC, relapse (viral and clinical) and HBsAg loss Q5: Renal function, hypophosphatemia and bone density Q6: Resistance, flare/decompensation and HBeAg loss Q7: Clinical outcomes: Cirrhosis, decompensated liver disease, HCC and death Study design RCT and controlled observational studies Acute HBV infection, children and pregnant women, HIV (+), HCV (+) or HDV (+) persons or other special populations Exclusions such as hemodialysis, transplant, and treatment failure populations. Co treatment with steroids and uncontrolled studies. Supplemental Table 2: Detailed Search Strategy: Ovid Database(s): Embase 1988 to 2014 Week 37, Ovid MEDLINE(R) In-Process & Other Non- Indexed Citations and Ovid MEDLINE(R) 1946 to Present, EBM Reviews - Cochrane Central Register of Controlled Trials August 2014, EBM Reviews - Cochrane Database of Systematic Reviews 2005 to July 2014 Search Strategy: # Searches Results 1 exp Hepatitis B/dt 26410 ("hepatitis B" or "serum hepatitis" or "hippie hepatitis" or "injection hepatitis" or 2 178548 "hepatitis type B").mp. -

(12) United States Patent (10) Patent No.: US 9.468,689 B2 Zeng Et Al
USOO9468689B2 (12) United States Patent (10) Patent No.: US 9.468,689 B2 Zeng et al. (45) Date of Patent: *Oct. 18, 2016 (54) ULTRAFILTRATION CONCENTRATION OF (56) References Cited ALLOTYPE SELECTED ANTIBODES FOR SMALL-VOLUME ADMINISTRATION U.S. PATENT DOCUMENTS 5,429,746 A 7/1995 Shadle et al. (71) Applicant: Immunomedics, Inc., Morris Plains, NJ 5,789,554 A 8/1998 Leung et al. (US) 6,171,586 B1 1/2001 Lam et al. 6,187,287 B1 2/2001 Leung et al. (72) Inventors: Li Zeng, Edison, NJ (US); Rohini 6,252,055 B1 6/2001 Relton Mitra, Brigdewater, NJ (US); Edmund 6,676,924 B2 1/2004 Hansen et al. 6,870,034 B2 3/2005 Breece et al. A. Rossi, Woodland Park, NJ (US); 6,893,639 B2 5/2005 Levy et al. Hans J. Hansen, Picayune, MS (US); 6,991,790 B1 1/2006 Lam et al. David M. Goldenberg, Mendham, NJ 7,038,017 B2 5, 2006 Rinderknecht et al. (US) 7,074,403 B1 7/2006 Goldenberg et al. 7,109,304 B2 9, 2006 Hansen et al. 7,138,496 B2 11/2006 Hua et al. (73) Assignee: Immunomedics, Inc., Morris Plains, NJ 7,151,164 B2 * 12/2006 Hansen et al. ............. 530,387.3 (US) 7,238,785 B2 7/2007 Govindan et al. 7,251,164 B2 7/2007 Okhonin et al. (*) Notice: Subject to any disclaimer, the term of this 7.282,567 B2 10/2007 Goldenberg et al. patent is extended or adjusted under 35 7,300,655 B2 11/2007 Hansen et al. -

WO 2015/028850 Al 5 March 2015 (05.03.2015) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2015/028850 Al 5 March 2015 (05.03.2015) P O P C T (51) International Patent Classification: AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, C07D 519/00 (2006.01) A61P 39/00 (2006.01) BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, C07D 487/04 (2006.01) A61P 35/00 (2006.01) DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, A61K 31/5517 (2006.01) A61P 37/00 (2006.01) HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KN, KP, KR, A61K 47/48 (2006.01) KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, (21) International Application Number: OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, PCT/IB2013/058229 SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, (22) International Filing Date: TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, 2 September 2013 (02.09.2013) ZW. (25) Filing Language: English (84) Designated States (unless otherwise indicated, for every kind of regional protection available): ARIPO (BW, GH, (26) Publication Language: English GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, SZ, TZ, (71) Applicant: HANGZHOU DAC BIOTECH CO., LTD UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, TJ, [US/CN]; Room B2001-B2019, Building 2, No 452 Sixth TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, DK, Street, Hangzhou Economy Development Area, Hangzhou EE, ES, FI, FR, GB, GR, HR, HU, IE, IS, IT, LT, LU, LV, City, Zhejiang 310018 (CN). -

Tanibirumab (CUI C3490677) Add to Cart
5/17/2018 NCI Metathesaurus Contains Exact Match Begins With Name Code Property Relationship Source ALL Advanced Search NCIm Version: 201706 Version 2.8 (using LexEVS 6.5) Home | NCIt Hierarchy | Sources | Help Suggest changes to this concept Tanibirumab (CUI C3490677) Add to Cart Table of Contents Terms & Properties Synonym Details Relationships By Source Terms & Properties Concept Unique Identifier (CUI): C3490677 NCI Thesaurus Code: C102877 (see NCI Thesaurus info) Semantic Type: Immunologic Factor Semantic Type: Amino Acid, Peptide, or Protein Semantic Type: Pharmacologic Substance NCIt Definition: A fully human monoclonal antibody targeting the vascular endothelial growth factor receptor 2 (VEGFR2), with potential antiangiogenic activity. Upon administration, tanibirumab specifically binds to VEGFR2, thereby preventing the binding of its ligand VEGF. This may result in the inhibition of tumor angiogenesis and a decrease in tumor nutrient supply. VEGFR2 is a pro-angiogenic growth factor receptor tyrosine kinase expressed by endothelial cells, while VEGF is overexpressed in many tumors and is correlated to tumor progression. PDQ Definition: A fully human monoclonal antibody targeting the vascular endothelial growth factor receptor 2 (VEGFR2), with potential antiangiogenic activity. Upon administration, tanibirumab specifically binds to VEGFR2, thereby preventing the binding of its ligand VEGF. This may result in the inhibition of tumor angiogenesis and a decrease in tumor nutrient supply. VEGFR2 is a pro-angiogenic growth factor receptor -

(12) United States Patent (10) Patent No.: US 8,658,773 B2 Zeng Et Al
USOO8658.773B2 (12) United States Patent (10) Patent No.: US 8,658,773 B2 Zeng et al. (45) Date of Patent: Feb. 25, 2014 (54) ULTRAFILTRATION CONCENTRATION OF 7,635,473 B2 12/2009 Warne et al. ALLOTYPE SELECTED ANTIBODES FOR Z$59. R: $388 E. et f SMALL-VOLUMEADMINISTRATION 7,829,5254 B2 11/2010 FrevertaSC ( a. (75) Inventors: Li Zeng, Edison, NJ (US); Rohini 7,847,071 B2 12/2010 Bonnerjea et al. Mitra, Brigdewater, NJ (US); Edmund 2. R: 138. Sagital. A. Rossi, Woodland Park, NJ (US); 7.87622 B2 1/2011 Changet al. Hans J. Hansen, Picayune, MS (US); 7,901,680 B2 3/2011 Chang et al. David M. Goldenberg, Mendham, NJ 7,906,118 B2 3/2011 Chang et al. (US) 7,906,121 B2 3/2011 Chang et al. 7.919,087 B2 4/2011 Hansen et al. (73) Assignee: Immunomedics, Inc., Morris Plains, NJ 7.931,903 B2 4/2011 Hansen et al. (US) 2001/0014326 A1 8/2001 Andya et al. 2003,0004094 A1 1/2003 Ghose et al. (*) Notice: Subject to any disclaimer, the term of this 2004/0033228 A1 2/2004 Krause et al. patent is extended or adjusted under 35 2004/0033561 A1 2/2004 O'Keefe et al. U.S.C. 154(b) by 0 days. 2004/0208870 A1 10, 2004 Allan 2005.0053666 A1 3/2005 TZannis et al. (21) Appl. No.: 13/461,307 2005, 0118167 A1 6, 2005 Okada et al. 2006/0153846 A1* 7/2006 Krause et al. .............. 424,145.1 (22) Filed: May 1, 2012 2006, O193850 A1 8, 2006 Warne et al. -

Looking for Therapeutic Antibodies in Next Generation Sequencing Repositories
bioRxiv preprint doi: https://doi.org/10.1101/572958; this version posted March 10, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Title: Looking for Therapeutic Antibodies in Next Generation Sequencing Repositories. Authors: Konrad Krawczyk1*, Matthew Raybould2, Aleksandr Kovaltsuk2, Charlotte M. Deane2 1 NaturalAntibody, Hamburg, Germany 2 Oxford University Department of Statistics, Oxford, UK *Correspondence to [email protected] Abstract: Recently it has become possible to query the great diversity of natural antibody repertoires using Next Generation Sequencing (NGS). These methods are capable of producing millions of sequences in a single experiment. Here we compare Clinical Stage Therapeutic antibodies to the ~1b sequences from 60 independent sequencing studies in the Observed Antibody Space Database. Of the 242 post Phase I antibodies, we find 16 with sequence identity matches of 95% or better for both heavy and light chains. There are also 54 perfect matches to therapeutic CDR-H3 regions in the NGS outputs, suggesting a nontrivial amount of convergence between naturally observed sequences and those developed artificially. This has potential implications for both the discovery of antibody therapeutics and the legal protection of commercial antibodies. Introduction Antibodies are proteins in jawed vertebrates that recognize noxious molecules (antigens) for elimination. An organism expresses millions of diverse antibodies to increase the chances that some of them will be able to bind the foreign antigen, initiating the adaptive immune response. -

Modifications to the Harmonized Tariff Schedule of the United States To
U.S. International Trade Commission COMMISSIONERS Shara L. Aranoff, Chairman Daniel R. Pearson, Vice Chairman Deanna Tanner Okun Charlotte R. Lane Irving A. Williamson Dean A. Pinkert Address all communications to Secretary to the Commission United States International Trade Commission Washington, DC 20436 U.S. International Trade Commission Washington, DC 20436 www.usitc.gov Modifications to the Harmonized Tariff Schedule of the United States to Implement the Dominican Republic- Central America-United States Free Trade Agreement With Respect to Costa Rica Publication 4038 December 2008 (This page is intentionally blank) Pursuant to the letter of request from the United States Trade Representative of December 18, 2008, set forth in the Appendix hereto, and pursuant to section 1207(a) of the Omnibus Trade and Competitiveness Act, the Commission is publishing the following modifications to the Harmonized Tariff Schedule of the United States (HTS) to implement the Dominican Republic- Central America-United States Free Trade Agreement, as approved in the Dominican Republic-Central America- United States Free Trade Agreement Implementation Act, with respect to Costa Rica. (This page is intentionally blank) Annex I Effective with respect to goods that are entered, or withdrawn from warehouse for consumption, on or after January 1, 2009, the Harmonized Tariff Schedule of the United States (HTS) is modified as provided herein, with bracketed matter included to assist in the understanding of proclaimed modifications. The following supersedes matter now in the HTS. (1). General note 4 is modified as follows: (a). by deleting from subdivision (a) the following country from the enumeration of independent beneficiary developing countries: Costa Rica (b). -

The Two Tontti Tudiul Lui Hi Ha Unit
THETWO TONTTI USTUDIUL 20170267753A1 LUI HI HA UNIT ( 19) United States (12 ) Patent Application Publication (10 ) Pub. No. : US 2017 /0267753 A1 Ehrenpreis (43 ) Pub . Date : Sep . 21 , 2017 ( 54 ) COMBINATION THERAPY FOR (52 ) U .S . CI. CO - ADMINISTRATION OF MONOCLONAL CPC .. .. CO7K 16 / 241 ( 2013 .01 ) ; A61K 39 / 3955 ANTIBODIES ( 2013 .01 ) ; A61K 31 /4706 ( 2013 .01 ) ; A61K 31 / 165 ( 2013 .01 ) ; CO7K 2317 /21 (2013 . 01 ) ; (71 ) Applicant: Eli D Ehrenpreis , Skokie , IL (US ) CO7K 2317/ 24 ( 2013. 01 ) ; A61K 2039/ 505 ( 2013 .01 ) (72 ) Inventor : Eli D Ehrenpreis, Skokie , IL (US ) (57 ) ABSTRACT Disclosed are methods for enhancing the efficacy of mono (21 ) Appl. No. : 15 /605 ,212 clonal antibody therapy , which entails co - administering a therapeutic monoclonal antibody , or a functional fragment (22 ) Filed : May 25 , 2017 thereof, and an effective amount of colchicine or hydroxy chloroquine , or a combination thereof, to a patient in need Related U . S . Application Data thereof . Also disclosed are methods of prolonging or increasing the time a monoclonal antibody remains in the (63 ) Continuation - in - part of application No . 14 / 947 , 193 , circulation of a patient, which entails co - administering a filed on Nov. 20 , 2015 . therapeutic monoclonal antibody , or a functional fragment ( 60 ) Provisional application No . 62/ 082, 682 , filed on Nov . of the monoclonal antibody , and an effective amount of 21 , 2014 . colchicine or hydroxychloroquine , or a combination thereof, to a patient in need thereof, wherein the time themonoclonal antibody remains in the circulation ( e . g . , blood serum ) of the Publication Classification patient is increased relative to the same regimen of admin (51 ) Int . -

WO 2016/176089 Al 3 November 2016 (03.11.2016) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2016/176089 Al 3 November 2016 (03.11.2016) P O P C T (51) International Patent Classification: BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, A01N 43/00 (2006.01) A61K 31/33 (2006.01) DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, (21) International Application Number: KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, PCT/US2016/028383 MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, (22) International Filing Date: PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, 20 April 2016 (20.04.2016) SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (25) Filing Language: English (84) Designated States (unless otherwise indicated, for every (26) Publication Language: English kind of regional protection available): ARIPO (BW, GH, (30) Priority Data: GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, 62/154,426 29 April 2015 (29.04.2015) US TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, TJ, TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, (71) Applicant: KARDIATONOS, INC. [US/US]; 4909 DK, EE, ES, FI, FR, GB, GR, HR, HU, IE, IS, IT, LT, LU, Lapeer Road, Metamora, Michigan 48455 (US). -

Μanagement of Patients with Hepatitis B and C Before and After Liver and Kidney Transplantation
Submit a Manuscript: http://www.wjgnet.com/esps/ World J Hepatol 2014 May 27; 6(5): 315-325 Help Desk: http://www.wjgnet.com/esps/helpdesk.aspx ISSN 1948-5182 (online) DOI: 10.4254/wjh.v6.i5.315 © 2014 Baishideng Publishing Group Inc. All rights reserved. REVIEW Μanagement of patients with hepatitis B and C before and after liver and kidney transplantation Chrysoula Pipili, Evangelos Cholongitas Chrysoula Pipili, Department of Nephrology, Laiki Merimna, © 2014 Baishideng Publishing Group Inc. All rights reserved. 17343 Athens, Greece th Evangelos Cholongitas, 4 Department of Internal Medicine, Key words: Liver transplantation; Kidney transplanta- Medical School of Aristotle University, Hippokration General tion; Hepatitis C; Hepatitis B; Recurrence Hospital of Thessaloniki, 54642 Thessaloniki, Greece Author contributions: Pipili C and Cholongitas E both contrib- Core tip: While nucleos(t)ide analogs (NAs) offer a be- uted to this paper. nign course in patients with hepatitis B virus before and Correspondence to: Evangelos Cholongitas, Assistant Pro- th after liver and renal transplantation, there is still scope fessor, 4 Department of Internal Medicine, Medical School of for improvement. The administration of high genetic Aristotle University, Hippokration General Hospital of Thessa- loniki, 49, Konstantinopoleos Street, 54642 Thessaloniki, barrier NAs such as entecavir or tenofovir pre-trans- Greece. [email protected] plant and the careful patient selection for hepatitis virus Telephone: +30-23-10892110 Fax: +30-23-10992940 immunoglobulin-free regimens post-transplant contrib- Received: December 3, 2013 Revised: March 10, 2014 ute to improved medical care and facilitate its provision Accepted: April 16, 2014 from a practical standpoint. Concordantly, attention has Published online: May 27, 2014 turned to new treatment strategies regarding hepatitis C virus recurrence after liver and renal transplantation. -

Advice Concerning the Addition of Certain Pharmaceutical Products
U.S. International Trade Commission COMMISSIONERS Daniel R. Pearson, Chairman Shara L. Aranoff, Vice Chairman Jennifer A. Hillman Stephen Koplan Deanna Tanner Okun Charlotte R. Lane Robert A. Rogowsky Director of Operations Karen Laney-Cummings Director of Industries Address all communications to Secretary to the Commission United States International Trade Commission Washington, DC 20436 U.S. International Trade Commission Washington, DC 20436 www.usitc.gov Advice Concerning the Addition of Certain Pharmaceutical Products and Chemical Intermediates to the Pharmaceutical Appendix to the Harmonized Tariff Schedule of the United States Investigation No. 332--476 Publication 3883 September 2006 This report was prepared principally by Office of Industries Philip Stone, Project Leader With assistance from Elizabeth R. Nesbitt Primary Reviewers David G. Michels, Office of Tariff Affairs and Trde Agreements, John Benedetto, and Nannette Christ, Office of Economics Administrative Support Brenda F. Carroll Under the direction of Dennis Rapkins, Chief Chemicals and Textiles Division ABSTRACT Under the Pharmaceutical Zero-for-Zero Initiative, which entered into force in 1995, the United States and its major trading partners eliminated tariffs on many pharmaceuticals, their derivatives, and certain chemical intermediates used to make pharmaceuticals. The U.S. list of pharmaceutical products and chemical intermediates eligible for duty-free treatment under the agreement is given in the Pharmaceutical Appendix to the Harmonized Tariff Schedule of the United States. The Pharmaceutical Appendix is periodically updated to provide duty relief for additional such products, including newly developed pharmaceuticals. This report provides advice on the third update to the agreement, in which approximately 1,300 products are proposed to receive duty-free treatment.