Phylogenomics Using Low-Depth Whole Genome Sequencing: a Case Study with the Olive Tribe
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Forestiera Lace Bugs Leptoypha Elliptica (From Wheeler, 2002) (No Common Names) Leptoypha Elliptica Leptoypha Ilicis Contributor: Alfred G
Forestiera Lace Bugs Leptoypha elliptica (from Wheeler, 2002) (No common names) Leptoypha elliptica Leptoypha ilicis Contributor: Alfred G. Wheeler DESCRIPTION Taxonomy and Basic Description Both adults and nymphs of the family Tingidae are strictly phytophagous. Lace bugs are mostly host restricted, developing on plants of only one genus or several related genera. They are found mainly on woody plants, the nymphs feeding on lower surfaces of leaves. Their feeding on mesophyll tissues usually results in a bleached or chlorotic appearance of the upper leaf surface and dark spots of excrement on the lower surface. Some lace bugs feed on herbaceous vascular plants and species of one genus (Acalypta) feed mainly on mosses. Lace bugs associated with small trees and shrubs of the genus Forestiera (olive family; Oleaceae) include two morphologically similar species of Leptoypha. McAtee described L. elliptica from Texas in 1917; L. ilicis was described in 1919 from Georgia by Drake. Only recently was the specific name of the latter species shown to be a Leptoypha ilicis (from Wheeler, 2002) misnomer. This lace bug was named ilicis because of its assumed host association: Ilex sp. Its actual hosts are species of Forestiera, the plant mentioned in the original description having been misidentified. Forestiera species bear a superficial resemblance to certain species of Ilex (holly; family Aquifoliaceae). Leptoypha species are small (about 3 mm long or 0.12 inches), elongate or narrowly oblong, and straw yellow to reddish brown. A hood (prothoracic covering of the head) and paranota (lateral expansions of pronotum), which characterize many lace bugs, are lacking. Also absent is the translucent lacy appearance of the forewings (hemelytra) that is typical of the Tingidae; elevations of the forewings also are absent. -

Brooklyn, Cloudland, Melsonby (Gaarraay)
BUSH BLITZ SPECIES DISCOVERY PROGRAM Brooklyn, Cloudland, Melsonby (Gaarraay) Nature Refuges Eubenangee Swamp, Hann Tableland, Melsonby (Gaarraay) National Parks Upper Bridge Creek Queensland 29 April–27 May · 26–27 July 2010 Australian Biological Resources Study What is Contents Bush Blitz? Bush Blitz is a four-year, What is Bush Blitz? 2 multi-million dollar Abbreviations 2 partnership between the Summary 3 Australian Government, Introduction 4 BHP Billiton and Earthwatch Reserves Overview 6 Australia to document plants Methods 11 and animals in selected properties across Australia’s Results 14 National Reserve System. Discussion 17 Appendix A: Species Lists 31 Fauna 32 This innovative partnership Vertebrates 32 harnesses the expertise of many Invertebrates 50 of Australia’s top scientists from Flora 62 museums, herbaria, universities, Appendix B: Threatened Species 107 and other institutions and Fauna 108 organisations across the country. Flora 111 Appendix C: Exotic and Pest Species 113 Fauna 114 Flora 115 Glossary 119 Abbreviations ANHAT Australian Natural Heritage Assessment Tool EPBC Act Environment Protection and Biodiversity Conservation Act 1999 (Commonwealth) NCA Nature Conservation Act 1992 (Queensland) NRS National Reserve System 2 Bush Blitz survey report Summary A Bush Blitz survey was conducted in the Cape Exotic vertebrate pests were not a focus York Peninsula, Einasleigh Uplands and Wet of this Bush Blitz, however the Cane Toad Tropics bioregions of Queensland during April, (Rhinella marina) was recorded in both Cloudland May and July 2010. Results include 1,186 species Nature Refuge and Hann Tableland National added to those known across the reserves. Of Park. Only one exotic invertebrate species was these, 36 are putative species new to science, recorded, the Spiked Awlsnail (Allopeas clavulinus) including 24 species of true bug, 9 species of in Cloudland Nature Refuge. -

(Rattus Spp. and Mus Musculus) in The
CHAPTER SIX: CONCLUSIONS Aaron B. Shiels Department of Botany University of Hawaii at Manoa 3190 Maile Way Honolulu, HI. 96822 173 Along with humans, introduced rats (Rattus rattus, R. norvegicus, and R. exulans) and mice (Mus musculus) are among the most invasive and widely distributed mammals on the planet; they occur on more than 80% of the world‘s islands groups (Atkinson 1985; Towns 2009). By incorporating modern technology, such as aerial broadcast of rodenticides, the number of islands where invasive rodents can be successfully removed has recently increased (Howald et al. 2007). However, successful rat and mouse eradication on relatively large (> 5000 ha) or human-inhabited islands such as the main Hawaiian Islands rarely occurs (Howald et al. 2007) despite large sums of money and research efforts annually to combat invasive rodent problems (see Chapter 1 section ―Rat history in Hawaii‖; Tobin et al. 1990). Therefore, it is highly unlikely that invasive rats and mice will be eradicated from relatively large, human-occupied islands such as Oahu in the near or distant future (Howald et al. 2007); and accepting this may be a first step towards increasing the likelihood of native species conservation in archipelagos like Hawaii where introduced rodents have established. Determining which invasive rodent species are present at a given site is important because the risks that some rodent species pose to particular (prey) species and/or habitats differ from those posed by other rodent species. Two sympatric species cannot occupy the same niche indefinitely, in a stable environment (Gause 1934), which may partly explain why some rodent species may not occur where others are present (Harper 2006). -

A New Application for Phylogenetic Marker Development Using Angiosperm Transcriptomes Author(S): Srikar Chamala, Nicolás García, Grant T
MarkerMiner 1.0: A New Application for Phylogenetic Marker Development Using Angiosperm Transcriptomes Author(s): Srikar Chamala, Nicolás García, Grant T. Godden, Vivek Krishnakumar, Ingrid E. Jordon- Thaden, Riet De Smet, W. Brad Barbazuk, Douglas E. Soltis, and Pamela S. Soltis Source: Applications in Plant Sciences, 3(4) Published By: Botanical Society of America DOI: http://dx.doi.org/10.3732/apps.1400115 URL: http://www.bioone.org/doi/full/10.3732/apps.1400115 BioOne (www.bioone.org) is a nonprofit, online aggregation of core research in the biological, ecological, and environmental sciences. BioOne provides a sustainable online platform for over 170 journals and books published by nonprofit societies, associations, museums, institutions, and presses. Your use of this PDF, the BioOne Web site, and all posted and associated content indicates your acceptance of BioOne’s Terms of Use, available at www.bioone.org/page/terms_of_use. Usage of BioOne content is strictly limited to personal, educational, and non-commercial use. Commercial inquiries or rights and permissions requests should be directed to the individual publisher as copyright holder. BioOne sees sustainable scholarly publishing as an inherently collaborative enterprise connecting authors, nonprofit publishers, academic institutions, research libraries, and research funders in the common goal of maximizing access to critical research. ApApplicatitionsons Applications in Plant Sciences 2015 3 ( 4 ): 1400115 inin PlPlant ScienSciencesces S OFTWARE NOTE M ARKERMINER 1.0: A NEW APPLICATION FOR PHYLOGENETIC 1 MARKER DEVELOPMENT USING ANGIOSPERM TRANSCRIPTOMES S RIKAR C HAMALA 2,12 , N ICOLÁS G ARCÍA 2,3,4 * , GRANT T . G ODDEN 2,3,5 * , V IVEK K RISHNAKUMAR 6 , I NGRID E. -

Recovery Plan for Tyoj5llllt . I-Bland Plants
Recovery Plan for tYOJ5llllt. i-bland Plants RECOVERY PLAN FOR MULTI-ISLAND PLANTS Published by U.S. Fish and Wildlife Service Portland, Oregon Approved: Date: / / As the Nation’s principal conservation agency, the Department of the Interior has responsibility for most ofour nationally owned public lands and natural resources. This includes fostering the wisest use ofour land and water resources, protecting our fish and wildlife, preserving the environmental and cultural values ofour national parks and historical places, and providing for the enjoyment of life through outdoor recreation. The Department assesses our energy and mineral resources and works to assure that their development is in the best interests ofall our people. The Department also has a major responsibility for American Indian reservation communities and for people who live in island Territories under U.S. administration. DISCLAIMER PAGE Recovery plans delineate reasonable actions that are believed to be required to recover and/or protect listed species. Plans are published by the U.S. Fish and Wildlife Service, sometimes prepared with the assistance ofrecovery teams, contractors, State agencies, and others. Objectives will be attained and any necessary funds made available subject to budgetary and other constraints affecting the parties involved, as well as the need to address other priorities. Costs indicated for task implementation and/or time for achievement ofrecovery are only estimates and are subject to change. Recovery plans do not necessarily represent the views nor the official positions or approval ofany individuals or agencies involved in the plan formulation, otherthan the U.S. Fish and Wildlife Service. They represent the official position ofthe U.S. -

Western Ghats & Sri Lanka Biodiversity Hotspot
Ecosystem Profile WESTERN GHATS & SRI LANKA BIODIVERSITY HOTSPOT WESTERN GHATS REGION FINAL VERSION MAY 2007 Prepared by: Kamal S. Bawa, Arundhati Das and Jagdish Krishnaswamy (Ashoka Trust for Research in Ecology & the Environment - ATREE) K. Ullas Karanth, N. Samba Kumar and Madhu Rao (Wildlife Conservation Society) in collaboration with: Praveen Bhargav, Wildlife First K.N. Ganeshaiah, University of Agricultural Sciences Srinivas V., Foundation for Ecological Research, Advocacy and Learning incorporating contributions from: Narayani Barve, ATREE Sham Davande, ATREE Balanchandra Hegde, Sahyadri Wildlife and Forest Conservation Trust N.M. Ishwar, Wildlife Institute of India Zafar-ul Islam, Indian Bird Conservation Network Niren Jain, Kudremukh Wildlife Foundation Jayant Kulkarni, Envirosearch S. Lele, Centre for Interdisciplinary Studies in Environment & Development M.D. Madhusudan, Nature Conservation Foundation Nandita Mahadev, University of Agricultural Sciences Kiran M.C., ATREE Prachi Mehta, Envirosearch Divya Mudappa, Nature Conservation Foundation Seema Purshothaman, ATREE Roopali Raghavan, ATREE T. R. Shankar Raman, Nature Conservation Foundation Sharmishta Sarkar, ATREE Mohammed Irfan Ullah, ATREE and with the technical support of: Conservation International-Center for Applied Biodiversity Science Assisted by the following experts and contributors: Rauf Ali Gladwin Joseph Uma Shaanker Rene Borges R. Kannan B. Siddharthan Jake Brunner Ajith Kumar C.S. Silori ii Milind Bunyan M.S.R. Murthy Mewa Singh Ravi Chellam Venkat Narayana H. Sudarshan B.A. Daniel T.S. Nayar R. Sukumar Ranjit Daniels Rohan Pethiyagoda R. Vasudeva Soubadra Devy Narendra Prasad K. Vasudevan P. Dharma Rajan M.K. Prasad Muthu Velautham P.S. Easa Asad Rahmani Arun Venkatraman Madhav Gadgil S.N. Rai Siddharth Yadav T. Ganesh Pratim Roy Santosh George P.S. -

Upland & Pollinator Plants Flip Book
Pictures Ilex opaca Callicarpa americana American Holly Beautyberry Prunus serotina Pteridium aquilinum Black Cherry Bracken Fern Descriptions Callicarpa americana Ilex opaca The American beautyberry is a shrub, 6-9 ft in height. Shoot This tree grows to be 35 to 50 ft tall and 15 to 25 ft wide. growth occurs throughout much of the season. It is It has a dense, pyramid-shaped crown. characterized by its attractive foliage and clusters of flowers or fruit around the leaf nodes. Leaves Leaves Alternate, simple and laceolate or elliptic in shape, 2-4 inches long and 1-1.5 inches wide. Medium to dark, Simple, opposite or subopposite, and deciduous. 3-5” long green in color, and shiny or flat. Leaf margins are usually and 1-3” wide, oval shaped, with an acute apex. Leaf bases spiny and will be flat or wavy. are tapered, margins are serrate. Surfaces are green and usually glabrous above, paler and pubescent below. Flowers Petioles are short and slender. Small, green or white in color, fragrant and found in Flowers clusters in the leaf axils. Perfect, in sessile clusters around the leaf nodes Bark Fruit The bark is light gray and may be covered by wart-like growths. Because the bark is very thin, it is easily A 4-pitted lavender-pink, magenta, or violet drupe, about 1/4 damaged. inch in diameter. Lasts long into winter and is eaten by a variety of birds. Interesting Facts Trunk The preserved berries of this tree were used by American Indians as decorative buttons. Historically, the Older stems have a thin protective, light-brown bark. -
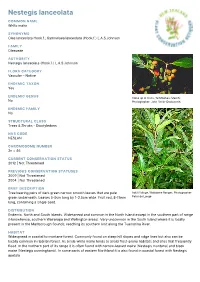
Nestegis Lanceolata
Nestegis lanceolata COMMON NAME White maire SYNONYMS Olea lanceolata Hook.f.; Gymnelaea lanceolata (Hook.f.) L.A.S.Johnson FAMILY Oleaceae AUTHORITY Nestegis lanceolata (Hook.f.) L.A.S.Johnson FLORA CATEGORY Vascular – Native ENDEMIC TAXON Yes ENDEMIC GENUS Close up of fruits, Te Moehau (March). No Photographer: John Smith-Dodsworth ENDEMIC FAMILY No STRUCTURAL CLASS Trees & Shrubs - Dicotyledons NVS CODE NESLAN CHROMOSOME NUMBER 2n = 46 CURRENT CONSERVATION STATUS 2012 | Not Threatened PREVIOUS CONSERVATION STATUSES 2009 | Not Threatened 2004 | Not Threatened BRIEF DESCRIPTION Tree bearing pairs of dark green narrow smooth leaves that are pale Adult foliage, Waitakere Ranges. Photographer: green underneath. Leaves 5-9cm long by 1-2.5cm wide. Fruit red, 8-11mm Peter de Lange long, containing a single seed. DISTRIBUTION Endemic. North and South Islands. Widespread and common in the North Island except in the southern part of range (Horowhenua, southern Wairarapa and Wellington areas). Very uncommon in the South Island where it is locally present in the Marlborough Sounds, reaching its southern limit along the Tuamarina River. HABITAT Widespread in coastal to montane forest. Commonly found on steep hill slopes and ridge lines but also can be locally common in riparian forest. As a rule white maire tends to avoid frost-prone habitats and sites that frequently flood. In the northern part of its range it is often found with narrow-leaved maire (Nestegis montana) and black maire (Nestegis cunninghamii). In some parts of eastern Northland it is also found in coastal forest with Nestegis apetala. FEATURES Stout gynodioecious spreading tree up to 20 m tall usually forming a domed canopy; trunk up to c. -

Chionanthus (Oleaceae) (Stapf) Kiew, Comb. Sagu, Padang Unique Among Being Ridged. Its Strongly Flattened Twigs, Long Petioles
BLUMEA 43 (1998) 471-477 Name changes for Malesian species of Chionanthus (Oleaceae) Ruth Kiew Singapore Botanic Gardens, Cluny Road, Singapore 259569 Summary New combinations under Chionanthus L. are made for Linociera beccarii, L. brassii, L. clementis, L. gigas, L. hahlii, L. kajewskii, L. nitida, L. remotinervia, L. riparia, L. rupicola, L. salicifolia, L. and L. Linociera is with C. L. sessiliflora stenura. cumingiana synonymous ramiflorus, novo- guineensis and L. ovalis with C. rupicolus, L. papuasica with C. sessiliflorus and L. pubipanicu- be lata with C. mala-elengi subsp. terniflorus. Linociera macrophylla sensu Whitmore proves to C. hahlii. Key words '. Malesia, Chionanthus,Oleaceae. Introduction The reduction of Linociera Sw. to Chionanthus L. (Steam, 1976) necessitates name changes to be made for species described inLinociera. In addition, as the family has been studied on a geographical rather than country basis, several species prove to be In for Vidal and two synonyms. some cases, notably a type Ledermanntypes, neotypes have had to be chosen because the holotype is no longer extant and isotypes have not been located. 1. Chionanthus beccarii (Stapf) Kiew, comb. nov. Linociera beccarii Stapf, Kew Bull. (1915) 115. —Type: Beccari PS 826 (holoK; isoL), Sumatra, Padang, Ayer Mancior. Distribution — Malesia: Sumatra (G. Leuser, G. Sagu, Padang and Asahan). — rather it is from Note Apparently a rare tree, known only the northern half of Sumatrawhere it grows in mountainforest between 360 and 1200 m. Its large fruit is unique among Malesian Chionanthus in being flattened laterally as well as being ridged. Its strongly flattened twigs, long petioles, narrowly obovate leaves with an acute apex, and rotund foliaceous bracts combine to make it a distinctive species. -

Price's Scrub State Park
Price’s Scrub State Park Advisory Group Draft Unit Management Plan STATE OF FLORIDA DEPARTMENT OF ENVIRONMENTAL PROTECTION Division of Recreation and Parks September 2018 TABLE OF CONTENTS INTRODUCTION ...................................................................................1 PURPOSE AND SIGNIFICANCE OF THE PARK ....................................... 1 Park Significance ................................................................................1 PURPOSE AND SCOPE OF THE PLAN..................................................... 2 MANAGEMENT PROGRAM OVERVIEW ................................................... 7 Management Authority and Responsibility .............................................. 7 Park Management Goals ...................................................................... 8 Management Coordination ................................................................... 9 Public Participation ..............................................................................9 Other Designations .............................................................................9 RESOURCE MANAGEMENT COMPONENT INTRODUCTION ................................................................................. 11 RESOURCE DESCRIPTION AND ASSESSMENT..................................... 12 Natural Resources ............................................................................. 12 Topography .................................................................................. 12 Geology ...................................................................................... -

Quercus ×Coutinhoi Samp. Discovered in Australia Charlie Buttigieg
XXX International Oaks The Journal of the International Oak Society …the hybrid oak that time forgot, oak-rod baskets, pros and cons of grafting… Issue No. 25/ 2014 / ISSN 1941-2061 1 International Oaks The Journal of the International Oak Society … the hybrid oak that time forgot, oak-rod baskets, pros and cons of grafting… Issue No. 25/ 2014 / ISSN 1941-2061 International Oak Society Officers and Board of Directors 2012-2015 Officers President Béatrice Chassé (France) Vice-President Charles Snyers d’Attenhoven (Belgium) Secretary Gert Fortgens (The Netherlands) Treasurer James E. Hitz (USA) Board of Directors Editorial Committee Membership Director Chairman Emily Griswold (USA) Béatrice Chassé Tour Director Members Shaun Haddock (France) Roderick Cameron International Oaks Allen Coombes Editor Béatrice Chassé Shaun Haddock Co-Editor Allen Coombes (Mexico) Eike Jablonski (Luxemburg) Oak News & Notes Ryan Russell Editor Ryan Russell (USA) Charles Snyers d’Attenhoven International Editor Roderick Cameron (Uruguay) Website Administrator Charles Snyers d’Attenhoven For contributions to International Oaks contact Béatrice Chassé [email protected] or [email protected] 0033553621353 Les Pouyouleix 24800 St.-Jory-de-Chalais France Author’s guidelines for submissions can be found at http://www.internationaloaksociety.org/content/author-guidelines-journal-ios © 2014 International Oak Society Text, figures, and photographs © of individual authors and photographers. Graphic design: Marie-Paule Thuaud / www.lecentrecreatifducoin.com Photos. Cover: Charles Snyers d’Attenhoven (Quercus macrocalyx Hickel & A. Camus); p. 6: Charles Snyers d’Attenhoven (Q. oxyodon Miq.); p. 7: Béatrice Chassé (Q. acerifolia (E.J. Palmer) Stoynoff & W. J. Hess); p. 9: Eike Jablonski (Q. ithaburensis subsp. -

SGAP Cairns Newsletter
SGAP Cairns Newsletter May 2018 Newsletter 179 Editor’s Note Society for Growing Australian Plants, Inc. Cairns Branch. www.sgapcairns.org.au You may have noticed this month’s newsletter is not as [email protected] “flashy” or to the standard we have come to expect each month from our newsletter editor, Stuart, that is because 2018 -2019 Committee he is taking a well earned holiday! However, what we President: Tony Roberts lack in pizzazz we have made up in content! Don has Vice President: Pauline Lawie kindly put together a report on our trip to Ella Bay (which Secretary: Sandy Perkins ([email protected]) was a great day out, btw) and the plant of the month Treasurer: Val Carnie Newsletter: including an interesting google translation. And of Stuart Worboys course, there are the details on our next excursion to ([email protected]) Emerald Creek Falls. Looking forward to seeing you all Webmaster: Tony Roberts in May. Sandy Perkins Excursion Report ELLA BAY (HEATH POINT ) Sunday 15 April 2018 By Don Lawie The beach and dune walk planned for 11 March was cancelled due to heavy rain, local flooding and road washouts. Indeed, damage to Ella Bay Road was so bad that it was closed at Heath Point, the southern arm of Ella Bay, when we arrived on 15 April. Nothing daunted, we set off along the beach but were soon blocked by sharp volcanic rocks so diverted to the road and walked up a steep hill then returned to the beach beyond the rock barrier. The aim of the day was to discover what plants – trees, shrubs, vines etc.- grew in the area with fruits that would conceivably be eaten by shipwrecked mariners who were not knowledgeable about their edibility or otherwise.