Supporting Information Materials and Methods
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplemental Information to Mammadova-Bach Et Al., “Laminin Α1 Orchestrates VEGFA Functions in the Ecosystem of Colorectal Carcinogenesis”
Supplemental information to Mammadova-Bach et al., “Laminin α1 orchestrates VEGFA functions in the ecosystem of colorectal carcinogenesis” Supplemental material and methods Cloning of the villin-LMα1 vector The plasmid pBS-villin-promoter containing the 3.5 Kb of the murine villin promoter, the first non coding exon, 5.5 kb of the first intron and 15 nucleotides of the second villin exon, was generated by S. Robine (Institut Curie, Paris, France). The EcoRI site in the multi cloning site was destroyed by fill in ligation with T4 polymerase according to the manufacturer`s instructions (New England Biolabs, Ozyme, Saint Quentin en Yvelines, France). Site directed mutagenesis (GeneEditor in vitro Site-Directed Mutagenesis system, Promega, Charbonnières-les-Bains, France) was then used to introduce a BsiWI site before the start codon of the villin coding sequence using the 5’ phosphorylated primer: 5’CCTTCTCCTCTAGGCTCGCGTACGATGACGTCGGACTTGCGG3’. A double strand annealed oligonucleotide, 5’GGCCGGACGCGTGAATTCGTCGACGC3’ and 5’GGCCGCGTCGACGAATTCACGC GTCC3’ containing restriction site for MluI, EcoRI and SalI were inserted in the NotI site (present in the multi cloning site), generating the plasmid pBS-villin-promoter-MES. The SV40 polyA region of the pEGFP plasmid (Clontech, Ozyme, Saint Quentin Yvelines, France) was amplified by PCR using primers 5’GGCGCCTCTAGATCATAATCAGCCATA3’ and 5’GGCGCCCTTAAGATACATTGATGAGTT3’ before subcloning into the pGEMTeasy vector (Promega, Charbonnières-les-Bains, France). After EcoRI digestion, the SV40 polyA fragment was purified with the NucleoSpin Extract II kit (Machery-Nagel, Hoerdt, France) and then subcloned into the EcoRI site of the plasmid pBS-villin-promoter-MES. Site directed mutagenesis was used to introduce a BsiWI site (5’ phosphorylated AGCGCAGGGAGCGGCGGCCGTACGATGCGCGGCAGCGGCACG3’) before the initiation codon and a MluI site (5’ phosphorylated 1 CCCGGGCCTGAGCCCTAAACGCGTGCCAGCCTCTGCCCTTGG3’) after the stop codon in the full length cDNA coding for the mouse LMα1 in the pCIS vector (kindly provided by P. -

The Activation of the Glucagon-Like Peptide-1 (GLP-1) Receptor by Peptide and Non-Peptide Ligands
The Activation of the Glucagon-Like Peptide-1 (GLP-1) Receptor by Peptide and Non-Peptide Ligands Clare Louise Wishart Submitted in accordance with the requirements for the degree of Doctor of Philosophy of Science University of Leeds School of Biomedical Sciences Faculty of Biological Sciences September 2013 I Intellectual Property and Publication Statements The candidate confirms that the work submitted is her own and that appropriate credit has been given where reference has been made to the work of others. This copy has been supplied on the understanding that it is copyright material and that no quotation from the thesis may be published without proper acknowledgement. The right of Clare Louise Wishart to be identified as Author of this work has been asserted by her in accordance with the Copyright, Designs and Patents Act 1988. © 2013 The University of Leeds and Clare Louise Wishart. II Acknowledgments Firstly I would like to offer my sincerest thanks and gratitude to my supervisor, Dr. Dan Donnelly, who has been nothing but encouraging and engaging from day one. I have thoroughly enjoyed every moment of working alongside him and learning from his guidance and wisdom. My thanks go to my academic assessor Professor Paul Milner whom I have known for several years, and during my time at the University of Leeds he has offered me invaluable advice and inspiration. Additionally I would like to thank my academic project advisor Dr. Michael Harrison for his friendship, help and advice. I would like to thank Dr. Rosalind Mann and Dr. Elsayed Nasr for welcoming me into the lab as a new PhD student and sharing their experimental techniques with me, these techniques have helped me no end in my time as a research student. -

Small Cell Ovarian Carcinoma: Genomic Stability and Responsiveness to Therapeutics
Gamwell et al. Orphanet Journal of Rare Diseases 2013, 8:33 http://www.ojrd.com/content/8/1/33 RESEARCH Open Access Small cell ovarian carcinoma: genomic stability and responsiveness to therapeutics Lisa F Gamwell1,2, Karen Gambaro3, Maria Merziotis2, Colleen Crane2, Suzanna L Arcand4, Valerie Bourada1,2, Christopher Davis2, Jeremy A Squire6, David G Huntsman7,8, Patricia N Tonin3,4,5 and Barbara C Vanderhyden1,2* Abstract Background: The biology of small cell ovarian carcinoma of the hypercalcemic type (SCCOHT), which is a rare and aggressive form of ovarian cancer, is poorly understood. Tumourigenicity, in vitro growth characteristics, genetic and genomic anomalies, and sensitivity to standard and novel chemotherapeutic treatments were investigated in the unique SCCOHT cell line, BIN-67, to provide further insight in the biology of this rare type of ovarian cancer. Method: The tumourigenic potential of BIN-67 cells was determined and the tumours formed in a xenograft model was compared to human SCCOHT. DNA sequencing, spectral karyotyping and high density SNP array analysis was performed. The sensitivity of the BIN-67 cells to standard chemotherapeutic agents and to vesicular stomatitis virus (VSV) and the JX-594 vaccinia virus was tested. Results: BIN-67 cells were capable of forming spheroids in hanging drop cultures. When xenografted into immunodeficient mice, BIN-67 cells developed into tumours that reflected the hypercalcemia and histology of human SCCOHT, notably intense expression of WT-1 and vimentin, and lack of expression of inhibin. Somatic mutations in TP53 and the most common activating mutations in KRAS and BRAF were not found in BIN-67 cells by DNA sequencing. -

Peking University-Juntendo University Joint Symposium on Cancer Research and Treatment ADAM28 (A Disintegrin and Metalloproteinase 28) in Cancer Cell Proliferation and Progression
Whatʼs New from Juntendo University, Tokyo Juntendo Medical Journal 2017. 63(5), 322-325 Peking University - Juntendo University Joint Symposium on Cancer Research and Treatment ADAM28 (a Disintegrin and Metalloproteinase 28) in Cancer Cell Proliferation and Progression YASUNORI OKADA* *Department of Pathophysiology for Locomotive and Neoplastic Diseases, Juntendo University Graduate School of Medicine, Tokyo, Japan A disintegrinandmetalloproteinase 28 (ADAM28) is overexpressedpredominantlyby carcinoma cells in more than 70% of the non-small cell lung carcinomas, showing positive correlations with carcinoma cell proliferation and metastasis. ADAM28 cleaves insulin-like growth factor binding protein-3 (IGFBP-3) in the IGF-I/IGFBP-3 complex, leading to stimulation of cell proliferation by intact IGF-I released from the complex. ADAM28 also degrades von Willebrand factor (VWF), which induces apoptosis in human carcinoma cell lines with negligible ADAM28 expression, andthe VWF digestionby ADAM28-expressing carcinoma cells facilitates them to escape from VWF-induced apoptosis, resulting in promotion of metastasis. We have developed human antibodies against ADAM28 andshown that one of them significantly inhibits tumor growth andmetastasis using lung adenocarcinoma cells. Our data suggest that ADAM28 may be a new molecular target for therapy of the patients with ADAM28-expressing non-small cell lung carcinoma. Key words: a disintegrin and metalloproteinase 28 (ADAM28), cell proliferation, invasion, metastasis, human antibody inhibitor Introduction human cancers 2). However, development of the synthetic inhibitors of MMPs andtheir application Cancer cell proliferation andprogression are for treatment of the cancer patients failed 3). modulated by proteolytic cleavage of tissue micro- On the other hand, members of the ADAM (a environmental factors such as extracellular matrix disintegrin and metalloproteinase) gene family, (ECM), growth factors andcytokines, receptors another family belonging to the metzincin gene andcell adhesionmolecules. -
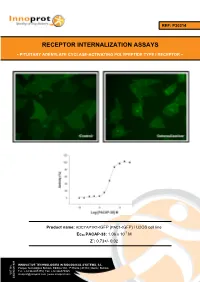
Receptor Internalization Assays
REF: P30214 RECEPTOR INTERNALIZATION ASSAYS - PITUITARY ADENYLATE CYCLASE-ACTIVATING POLYPEPTIDE TYPE I RECEPTOR - Product name: ADCYAP1R1-tGFP (PAC1-tGFP) / U2OS cell line -7 Ec50 PACAP-38: 1.06 x 10 M Z´: 0.73+/- 0.02 INNOVATIVE TECHNOLOGIES IN BIOLOGICAL SYSTEMS, S.L. Parque Tecnológico Bizkaia, Edifício 502, 1ª Planta | 48160 | Derio | Bizkaia Tel.: +34 944005355 | Fax: +34 946579925 VAT No. [email protected] | www.innoprot.com ESB95481909 Product Name: ADCYAP1R1-tGFP_U2OS Reference: P30214 Rep. Official Full Name: Pituitary adenylate cyclase- activating polypeptide type I receptor DNA Accession Number: Gene Bank AY366498 Host Cell: U2OS References: P30214: 2 vials of 3 x 106 proliferative cells P30214-DA: 1 vial of 2 x 106 division-arrested cells Storage: Liquid Nitrogen Assay Briefly description About ADCYAP1R1 Each vial of ADCYAP1R1 Internalization Assay Pituitary adenylate cyclase-activating Cell Line contains U2OS cells stably expressing polypeptide type I receptor, also known as human Pituitary adenylate cyclase-activating PAC1 is a protein that in humans is encoded by polypeptide type I receptor tagged in the N- the ADCYAP1R1 gene. ADCYAP1R1 is a terminus with tGFP protein. membrane-associated protein and shares significant homology with members of the Innoprot’s ADCYAP1R1-tGFP Internalization glucagon/secretin receptor family. This receptor Assay Cell Line has been designed to assay binds pituitary adenylate cyclase activating potential agonists/ antagonists against peptide (PACAP) mediating several biological ADCYAP1R1, modulating its activation and the activities and it is positively coupled to following redistribution process inside the cells. adenylate cyclase. This cell line will allow the image analysis of the stimuli induced by the compounds. -

An Animal Model with a Cardiomyocyte-Specific Deletion of Estrogen Receptor Alpha: Functional, Metabolic, and Differential Netwo
Washington University School of Medicine Digital Commons@Becker Open Access Publications 2014 An animal model with a cardiomyocyte-specific deletion of estrogen receptor alpha: Functional, metabolic, and differential network analysis Sriram Devanathan Washington University School of Medicine in St. Louis Timothy Whitehead Washington University School of Medicine in St. Louis George G. Schweitzer Washington University School of Medicine in St. Louis Nicole Fettig Washington University School of Medicine in St. Louis Attila Kovacs Washington University School of Medicine in St. Louis See next page for additional authors Follow this and additional works at: https://digitalcommons.wustl.edu/open_access_pubs Recommended Citation Devanathan, Sriram; Whitehead, Timothy; Schweitzer, George G.; Fettig, Nicole; Kovacs, Attila; Korach, Kenneth S.; Finck, Brian N.; and Shoghi, Kooresh I., ,"An animal model with a cardiomyocyte-specific deletion of estrogen receptor alpha: Functional, metabolic, and differential network analysis." PLoS One.9,7. e101900. (2014). https://digitalcommons.wustl.edu/open_access_pubs/3326 This Open Access Publication is brought to you for free and open access by Digital Commons@Becker. It has been accepted for inclusion in Open Access Publications by an authorized administrator of Digital Commons@Becker. For more information, please contact [email protected]. Authors Sriram Devanathan, Timothy Whitehead, George G. Schweitzer, Nicole Fettig, Attila Kovacs, Kenneth S. Korach, Brian N. Finck, and Kooresh I. Shoghi This open access publication is available at Digital Commons@Becker: https://digitalcommons.wustl.edu/open_access_pubs/3326 An Animal Model with a Cardiomyocyte-Specific Deletion of Estrogen Receptor Alpha: Functional, Metabolic, and Differential Network Analysis Sriram Devanathan1, Timothy Whitehead1, George G. Schweitzer2, Nicole Fettig1, Attila Kovacs3, Kenneth S. -

The Role and Mechanisms of Action of Micrornas in Cancer Drug Resistance Wengong Si1,2,3, Jiaying Shen4, Huilin Zheng1,5 and Weimin Fan1,6*
Si et al. Clinical Epigenetics (2019) 11:25 https://doi.org/10.1186/s13148-018-0587-8 REVIEW Open Access The role and mechanisms of action of microRNAs in cancer drug resistance Wengong Si1,2,3, Jiaying Shen4, Huilin Zheng1,5 and Weimin Fan1,6* Abstract MicroRNAs (miRNAs) are small non-coding RNAs with a length of about 19–25 nt, which can regulate various target genes and are thus involved in the regulation of a variety of biological and pathological processes, including the formation and development of cancer. Drug resistance in cancer chemotherapy is one of the main obstacles to curing this malignant disease. Statistical data indicate that over 90% of the mortality of patients with cancer is related to drug resistance. Drug resistance of cancer chemotherapy can be caused by many mechanisms, such as decreased antitumor drug uptake, modified drug targets, altered cell cycle checkpoints, or increased DNA damage repair, among others. In recent years, many studies have shown that miRNAs are involved in the drug resistance of tumor cells by targeting drug-resistance-related genes or influencing genes related to cell proliferation, cell cycle, and apoptosis. A single miRNA often targets a number of genes, and its regulatory effect is tissue-specific. In this review, we emphasize the miRNAs that are involved in the regulation of drug resistance among different cancers and probe the mechanisms of the deregulated expression of miRNAs. The molecular targets of miRNAs and their underlying signaling pathways are also explored comprehensively. A holistic understanding of the functions of miRNAs in drug resistance will help us develop better strategies to regulate them efficiently and will finally pave the way toward better translation of miRNAs into clinics, developing them into a promising approach in cancer therapy. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

ADAM10 Site-Dependent Biology: Keeping Control of a Pervasive Protease
International Journal of Molecular Sciences Review ADAM10 Site-Dependent Biology: Keeping Control of a Pervasive Protease Francesca Tosetti 1,* , Massimo Alessio 2, Alessandro Poggi 1,† and Maria Raffaella Zocchi 3,† 1 Molecular Oncology and Angiogenesis Unit, IRCCS Ospedale Policlinico S. Martino Largo R. Benzi 10, 16132 Genoa, Italy; [email protected] 2 Proteome Biochemistry, IRCCS San Raffaele Scientific Institute, 20132 Milan, Italy; [email protected] 3 Division of Immunology, Transplants and Infectious Diseases, IRCCS San Raffaele Scientific Institute, 20132 Milan, Italy; [email protected] * Correspondence: [email protected] † These authors contributed equally to this work as last author. Abstract: Enzymes, once considered static molecular machines acting in defined spatial patterns and sites of action, move to different intra- and extracellular locations, changing their function. This topological regulation revealed a close cross-talk between proteases and signaling events involving post-translational modifications, membrane tyrosine kinase receptors and G-protein coupled recep- tors, motor proteins shuttling cargos in intracellular vesicles, and small-molecule messengers. Here, we highlight recent advances in our knowledge of regulation and function of A Disintegrin And Metalloproteinase (ADAM) endopeptidases at specific subcellular sites, or in multimolecular com- plexes, with a special focus on ADAM10, and tumor necrosis factor-α convertase (TACE/ADAM17), since these two enzymes belong to the same family, share selected substrates and bioactivity. We will discuss some examples of ADAM10 activity modulated by changing partners and subcellular compartmentalization, with the underlying hypothesis that restraining protease activity by spatial Citation: Tosetti, F.; Alessio, M.; segregation is a complex and powerful regulatory tool. -

Supplementary Table 1: Adhesion Genes Data Set
Supplementary Table 1: Adhesion genes data set PROBE Entrez Gene ID Celera Gene ID Gene_Symbol Gene_Name 160832 1 hCG201364.3 A1BG alpha-1-B glycoprotein 223658 1 hCG201364.3 A1BG alpha-1-B glycoprotein 212988 102 hCG40040.3 ADAM10 ADAM metallopeptidase domain 10 133411 4185 hCG28232.2 ADAM11 ADAM metallopeptidase domain 11 110695 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 195222 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 165344 8751 hCG20021.3 ADAM15 ADAM metallopeptidase domain 15 (metargidin) 189065 6868 null ADAM17 ADAM metallopeptidase domain 17 (tumor necrosis factor, alpha, converting enzyme) 108119 8728 hCG15398.4 ADAM19 ADAM metallopeptidase domain 19 (meltrin beta) 117763 8748 hCG20675.3 ADAM20 ADAM metallopeptidase domain 20 126448 8747 hCG1785634.2 ADAM21 ADAM metallopeptidase domain 21 208981 8747 hCG1785634.2|hCG2042897 ADAM21 ADAM metallopeptidase domain 21 180903 53616 hCG17212.4 ADAM22 ADAM metallopeptidase domain 22 177272 8745 hCG1811623.1 ADAM23 ADAM metallopeptidase domain 23 102384 10863 hCG1818505.1 ADAM28 ADAM metallopeptidase domain 28 119968 11086 hCG1786734.2 ADAM29 ADAM metallopeptidase domain 29 205542 11085 hCG1997196.1 ADAM30 ADAM metallopeptidase domain 30 148417 80332 hCG39255.4 ADAM33 ADAM metallopeptidase domain 33 140492 8756 hCG1789002.2 ADAM7 ADAM metallopeptidase domain 7 122603 101 hCG1816947.1 ADAM8 ADAM metallopeptidase domain 8 183965 8754 hCG1996391 ADAM9 ADAM metallopeptidase domain 9 (meltrin gamma) 129974 27299 hCG15447.3 ADAMDEC1 ADAM-like, -

An Investigation of ADAM-Like Decysin 1 in Macrophage-Mediated Inflammation and Crohn’S Disease
An Investigation of ADAM-like Decysin 1 in Macrophage-mediated Inflammation and Crohn’s Disease By Nuala Roisin O’Shea A thesis submitted to UCL for the degree of Doctor of Philosophy Division of Medicine Declaration I, Nuala Roisin O’Shea, confirm that the work presented in this thesis is my own. Where information has been derived from other sources, I confirm that this has been indicated in the thesis. 2 Abstract Crohn’s disease (CD) is now recognised as a defective host response to bacteria in genetically susceptible individuals. The role of innate immunity and impaired bacterial clearance are widely accepted. In this thesis the role of ADAM-like, Decysin-1 (ADAMDEC1) in macrophage-mediated inflammation and gut mucosal immunity is explored. Using transcriptomic analysis of monocyte derived macrophages (MDM) ADAMDEC1 was identified as grossly under expressed in a subset of patients with CD. ADAMDEC1 was found to be highly selective to the intestine, peripheral blood monocyte-derived and lamina propria macrophages. It was shown to be an inflammatory response gene, upregulated in response to bacterial antigens and inflammation. ADAMDEC1 was expressed in prenatal and germ free mice, demonstrating exposure to a bacterial antigen is not a prerequisite for expression. Adamdec1 knock out mice were used to investigate the role of ADAMDEC1 in vivo. Adamdec1-/- mice displayed an increased susceptibility to dextran sodium sulphate (DSS), Citrobacter rodentium and Salmonella typhimurium induced colitis. In Adamdec1-/- mice, bacterial translocation and systemic infection were increased in bacterial models of colitis. These results suggest that individuals with grossly attenuated expression of ADAMDEC1 may be at an increased risk of developing intestinal inflammation as a consequence of an impaired ability to handle enteric bacterial pathogens. -

NATURAL KILLER CELLS, HYPOXIA, and EPIGENETIC REGULATION of HEMOCHORIAL PLACENTATION by Damayanti Chakraborty Submitted to the G
NATURAL KILLER CELLS, HYPOXIA, AND EPIGENETIC REGULATION OF HEMOCHORIAL PLACENTATION BY Damayanti Chakraborty Submitted to the graduate degree program in Pathology and Laboratory Medicine and the Graduate Faculty of the University of Kansas in partial fulfillment ofthe requirements for the degree of Doctor of Philosophy. ________________________________ Chair: Michael J. Soares, Ph.D. ________________________________ Jay Vivian, Ph.D. ________________________________ Patrick Fields, Ph.D. ________________________________ Soumen Paul, Ph.D. ________________________________ Michael Wolfe, Ph.D. ________________________________ Adam J. Krieg, Ph.D. Date Defended: 04/01/2013 The Dissertation Committee for Damayanti Chakraborty certifies that this is the approved version of the following dissertation: NATURAL KILLER CELLS, HYPOXIA, AND EPIGENETIC REGULATION OF HEMOCHORIAL PLACENTATION ________________________________ Chair: Michael J. Soares, Ph.D. Date approved: 04/01/2013 ii ABSTRACT During the establishment of pregnancy, uterine stromal cells differentiate into decidual cells and recruit natural killer (NK) cells. These NK cells are characterized by low cytotoxicity and distinct cytokine production. In rodent as well as in human pregnancy, the uterine NK cells peak in number around mid-gestation after which they decline. NK cells associate with uterine spiral arteries and are implicated in pregnancy associated vascular remodeling processes and potentially in modulating trophoblast invasion. Failure of trophoblast invasion and vascular remodeling has been shown to be associated with pathological conditions like preeclampsia syndrome, hypertension in mother and/or fetal growth restriction. We hypothesize that NK cells fundamentally contribute to the organization of the placentation site. In order to study the in vivo role of NK cells during pregnancy, gestation stage- specific NK cell depletion was performed in rats using anti asialo GM1 antibodies.