Cloning, Sequencing, Mapping, and Expression of the Gene
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Generated by SRI International Pathway Tools Version 25.0, Authors S
An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_000238675-HmpCyc: Bacillus smithii 7_3_47FAA Cellular Overview Connections between pathways are omitted for legibility. -

Yeast Genome Gazetteer P35-65
gazetteer Metabolism 35 tRNA modification mitochondrial transport amino-acid metabolism other tRNA-transcription activities vesicular transport (Golgi network, etc.) nitrogen and sulphur metabolism mRNA synthesis peroxisomal transport nucleotide metabolism mRNA processing (splicing) vacuolar transport phosphate metabolism mRNA processing (5’-end, 3’-end processing extracellular transport carbohydrate metabolism and mRNA degradation) cellular import lipid, fatty-acid and sterol metabolism other mRNA-transcription activities other intracellular-transport activities biosynthesis of vitamins, cofactors and RNA transport prosthetic groups other transcription activities Cellular organization and biogenesis 54 ionic homeostasis organization and biogenesis of cell wall and Protein synthesis 48 plasma membrane Energy 40 ribosomal proteins organization and biogenesis of glycolysis translation (initiation,elongation and cytoskeleton gluconeogenesis termination) organization and biogenesis of endoplasmic pentose-phosphate pathway translational control reticulum and Golgi tricarboxylic-acid pathway tRNA synthetases organization and biogenesis of chromosome respiration other protein-synthesis activities structure fermentation mitochondrial organization and biogenesis metabolism of energy reserves (glycogen Protein destination 49 peroxisomal organization and biogenesis and trehalose) protein folding and stabilization endosomal organization and biogenesis other energy-generation activities protein targeting, sorting and translocation vacuolar and lysosomal -

Letters to Nature
letters to nature Received 7 July; accepted 21 September 1998. 26. Tronrud, D. E. Conjugate-direction minimization: an improved method for the re®nement of macromolecules. Acta Crystallogr. A 48, 912±916 (1992). 1. Dalbey, R. E., Lively, M. O., Bron, S. & van Dijl, J. M. The chemistry and enzymology of the type 1 27. Wolfe, P. B., Wickner, W. & Goodman, J. M. Sequence of the leader peptidase gene of Escherichia coli signal peptidases. Protein Sci. 6, 1129±1138 (1997). and the orientation of leader peptidase in the bacterial envelope. J. Biol. Chem. 258, 12073±12080 2. Kuo, D. W. et al. Escherichia coli leader peptidase: production of an active form lacking a requirement (1983). for detergent and development of peptide substrates. Arch. Biochem. Biophys. 303, 274±280 (1993). 28. Kraulis, P.G. Molscript: a program to produce both detailed and schematic plots of protein structures. 3. Tschantz, W. R. et al. Characterization of a soluble, catalytically active form of Escherichia coli leader J. Appl. Crystallogr. 24, 946±950 (1991). peptidase: requirement of detergent or phospholipid for optimal activity. Biochemistry 34, 3935±3941 29. Nicholls, A., Sharp, K. A. & Honig, B. Protein folding and association: insights from the interfacial and (1995). the thermodynamic properties of hydrocarbons. Proteins Struct. Funct. Genet. 11, 281±296 (1991). 4. Allsop, A. E. et al.inAnti-Infectives, Recent Advances in Chemistry and Structure-Activity Relationships 30. Meritt, E. A. & Bacon, D. J. Raster3D: photorealistic molecular graphics. Methods Enzymol. 277, 505± (eds Bently, P. H. & O'Hanlon, P. J.) 61±72 (R. Soc. Chem., Cambridge, 1997). -

Uracil and Beta-Alanine Degradation in Saccharomyces Kluyveri - Discovery of a Novel Catabolic Pathway
Downloaded from orbit.dtu.dk on: Sep 30, 2021 Uracil and beta-alanine degradation in Saccharomyces Kluyveri - discovery of a novel catabolic pathway Andersen, Gorm Publication date: 2006 Document Version Publisher's PDF, also known as Version of record Link back to DTU Orbit Citation (APA): Andersen, G. (2006). Uracil and beta-alanine degradation in Saccharomyces Kluyveri - discovery of a novel catabolic pathway. Technical University of Denmark. General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. Users may download and print one copy of any publication from the public portal for the purpose of private study or research. You may not further distribute the material or use it for any profit-making activity or commercial gain You may freely distribute the URL identifying the publication in the public portal If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. Uracil and beta-alanine degradation in Saccharomyces kluyveri - Discovery of a novel catabolic pathway Ph.D. Thesis Gorm Andersen BioCentrum-DTU Technical University of Denmark May 2006 PREFACE The thesis presented here is a part of the requirements for the Ph.D. degree under the ”Chemistry and Biotechnology Programme” at the Technical University of Denmark (DTU). Since October 2002, I have been enrolled at BioCentrum-DTU. The study was financed by DTU and has been carried out under supervision of former Associate Professor at BioCentrum-DTU, (now Professor at Department of Cell and Organism Biology, Lund University, Sweden) Jure Piskur. -

Study on Yeast Enzymes Urc1p and Urc4p in a Novel Uracil Catabolic Pathway (URC)
Study on yeast enzymes Urc1p and Urc4p in a novel uracil catabolic pathway (URC) Dineshkumar Kandasamy Degree project in molecular biotechnology, 2012 Examensarbete i molekylär bioteknik 45 hp till masterexamen, 2012 Biology Education Centre, Uppsala University, and Department of Biology, Lund University Supervisors: Jure Piškur and Anna Rasmussen ABSTRACT Purine and pyrimidine bases are central precursors of DNA and RNA and their intracellular concentration is balanced by three pathways- de novo, salvage and catabolic pathways. Uracil catabolism pathway has been found in several bacteria and in some fungi (including yeast). Seven genes, URC1-7 have been found to be involved in this novel pathway. There are two “unknown genes” in the yeast Saccharomyces (Lachancea) kluyveri, namely URC1 and URC4, which play a central role in this pathway and their exact function remains a mystery. In this project, two S. kluyveri genes, URC1&URC4, were over-expressed in the bacterial system and successfully purified. Our preliminary functional assay showed that uridine monophosphate (UMP) is a likely substrate for Urc1p at pH 7, 25ºC. It was shown clearly that both uracil and uridine were not the substrate for Urc1p.We tried to phosphorylate chemically synthesized ribosylurea (RU) using Drosophila melanogaster deoxyribonucleoside kinase and compared the activity between phosphorylated and non- phosphorylated RU at different conditions. Phosphorylated ribosylurea seemed to be a likely substrate for Urc4p at pH 7, 37ºC. Keywords: Uridine monophosphate (UMP), ribosylurea (RU), uracil catabolism. Study on yeast enzymes Urc1p and Urc4p in a novel uracil catabolic pathway (URC) Popular science summary Dineshkumar Kandasamy Purines and pyrimidines are important constituent of nucleic acids and they are called building blocks of DNA and RNA. -

The Microbiota-Produced N-Formyl Peptide Fmlf Promotes Obesity-Induced Glucose
Page 1 of 230 Diabetes Title: The microbiota-produced N-formyl peptide fMLF promotes obesity-induced glucose intolerance Joshua Wollam1, Matthew Riopel1, Yong-Jiang Xu1,2, Andrew M. F. Johnson1, Jachelle M. Ofrecio1, Wei Ying1, Dalila El Ouarrat1, Luisa S. Chan3, Andrew W. Han3, Nadir A. Mahmood3, Caitlin N. Ryan3, Yun Sok Lee1, Jeramie D. Watrous1,2, Mahendra D. Chordia4, Dongfeng Pan4, Mohit Jain1,2, Jerrold M. Olefsky1 * Affiliations: 1 Division of Endocrinology & Metabolism, Department of Medicine, University of California, San Diego, La Jolla, California, USA. 2 Department of Pharmacology, University of California, San Diego, La Jolla, California, USA. 3 Second Genome, Inc., South San Francisco, California, USA. 4 Department of Radiology and Medical Imaging, University of Virginia, Charlottesville, VA, USA. * Correspondence to: 858-534-2230, [email protected] Word Count: 4749 Figures: 6 Supplemental Figures: 11 Supplemental Tables: 5 1 Diabetes Publish Ahead of Print, published online April 22, 2019 Diabetes Page 2 of 230 ABSTRACT The composition of the gastrointestinal (GI) microbiota and associated metabolites changes dramatically with diet and the development of obesity. Although many correlations have been described, specific mechanistic links between these changes and glucose homeostasis remain to be defined. Here we show that blood and intestinal levels of the microbiota-produced N-formyl peptide, formyl-methionyl-leucyl-phenylalanine (fMLF), are elevated in high fat diet (HFD)- induced obese mice. Genetic or pharmacological inhibition of the N-formyl peptide receptor Fpr1 leads to increased insulin levels and improved glucose tolerance, dependent upon glucagon- like peptide-1 (GLP-1). Obese Fpr1-knockout (Fpr1-KO) mice also display an altered microbiome, exemplifying the dynamic relationship between host metabolism and microbiota. -

Chemical Transformations Encoded by a Gene Cluster in Streptomyces Coelicolor Containing an Unusual Gtp Cyclohydrolase
Chemical Transformations Encoded by a Streptomyces coelicolor Gene Cluster with an Unusual GTP Cyclohydrolase Item Type text; Electronic Dissertation Authors Spoonamore, James Edward Publisher The University of Arizona. Rights Copyright © is held by the author. Digital access to this material is made possible by the University Libraries, University of Arizona. Further transmission, reproduction or presentation (such as public display or performance) of protected items is prohibited except with permission of the author. Download date 11/10/2021 06:44:52 Link to Item http://hdl.handle.net/10150/194825 CHEMICAL TRANSFORMATIONS ENCODED BY A GENE CLUSTER IN STREPTOMYCES COELICOLOR CONTAINING AN UNUSUAL GTP CYCLOHYDROLASE by James Edward Spoonamore ______________________________ A Dissertation Submitted to the Faculty of the DEPARTMENT OF BIOCHEMISTRY AND MOLECULAR BIOPHYSICS In Partial Fulfillment of the Requirements For the Degree of DOCTOR OF PHILOSOPHY In the Graduate College THE UNIVERSITY OF ARIZONA 2008 2 THE UNIVERSITY OF ARIZONA GRADUATE COLLEGE As members of the Dissertation Committee, we certify that we have read the dissertation prepared by James Edward Spoonamore entitled Chemical Transformations Encoded by a Gene Cluster in Streptomyces coelicolor Containing an Unusual GTP Cyclohydrolase and recommend that it be accepted as fulfilling the dissertation requirement for the Degree of Doctor of Philosophy _______________________________________________________________________ Date: April 16, 2008 Vahe Bandarian _______________________________________________________________________ -

Supplemental Table S1: Comparison of the Deleted Genes in the Genome-Reduced Strains
Supplemental Table S1: Comparison of the deleted genes in the genome-reduced strains Legend 1 Locus tag according to the reference genome sequence of B. subtilis 168 (NC_000964) Genes highlighted in blue have been deleted from the respective strains Genes highlighted in green have been inserted into the indicated strain, they are present in all following strains Regions highlighted in red could not be deleted as a unit Regions highlighted in orange were not deleted in the genome-reduced strains since their deletion resulted in severe growth defects Gene BSU_number 1 Function ∆6 IIG-Bs27-47-24 PG10 PS38 dnaA BSU00010 replication initiation protein dnaN BSU00020 DNA polymerase III (beta subunit), beta clamp yaaA BSU00030 unknown recF BSU00040 repair, recombination remB BSU00050 involved in the activation of biofilm matrix biosynthetic operons gyrB BSU00060 DNA-Gyrase (subunit B) gyrA BSU00070 DNA-Gyrase (subunit A) rrnO-16S- trnO-Ala- trnO-Ile- rrnO-23S- rrnO-5S yaaC BSU00080 unknown guaB BSU00090 IMP dehydrogenase dacA BSU00100 penicillin-binding protein 5*, D-alanyl-D-alanine carboxypeptidase pdxS BSU00110 pyridoxal-5'-phosphate synthase (synthase domain) pdxT BSU00120 pyridoxal-5'-phosphate synthase (glutaminase domain) serS BSU00130 seryl-tRNA-synthetase trnSL-Ser1 dck BSU00140 deoxyadenosin/deoxycytidine kinase dgk BSU00150 deoxyguanosine kinase yaaH BSU00160 general stress protein, survival of ethanol stress, SafA-dependent spore coat yaaI BSU00170 general stress protein, similar to isochorismatase yaaJ BSU00180 tRNA specific adenosine -

12) United States Patent (10
US007635572B2 (12) UnitedO States Patent (10) Patent No.: US 7,635,572 B2 Zhou et al. (45) Date of Patent: Dec. 22, 2009 (54) METHODS FOR CONDUCTING ASSAYS FOR 5,506,121 A 4/1996 Skerra et al. ENZYME ACTIVITY ON PROTEIN 5,510,270 A 4/1996 Fodor et al. MICROARRAYS 5,512,492 A 4/1996 Herron et al. 5,516,635 A 5/1996 Ekins et al. (75) Inventors: Fang X. Zhou, New Haven, CT (US); 5,532,128 A 7/1996 Eggers Barry Schweitzer, Cheshire, CT (US) 5,538,897 A 7/1996 Yates, III et al. s s 5,541,070 A 7/1996 Kauvar (73) Assignee: Life Technologies Corporation, .. S.E. al Carlsbad, CA (US) 5,585,069 A 12/1996 Zanzucchi et al. 5,585,639 A 12/1996 Dorsel et al. (*) Notice: Subject to any disclaimer, the term of this 5,593,838 A 1/1997 Zanzucchi et al. patent is extended or adjusted under 35 5,605,662 A 2f1997 Heller et al. U.S.C. 154(b) by 0 days. 5,620,850 A 4/1997 Bamdad et al. 5,624,711 A 4/1997 Sundberg et al. (21) Appl. No.: 10/865,431 5,627,369 A 5/1997 Vestal et al. 5,629,213 A 5/1997 Kornguth et al. (22) Filed: Jun. 9, 2004 (Continued) (65) Prior Publication Data FOREIGN PATENT DOCUMENTS US 2005/O118665 A1 Jun. 2, 2005 EP 596421 10, 1993 EP 0619321 12/1994 (51) Int. Cl. EP O664452 7, 1995 CI2O 1/50 (2006.01) EP O818467 1, 1998 (52) U.S. -

Global Response of Saccharomyces Cerevisiae to an Alkylating Agent
Proc. Natl. Acad. Sci. USA Vol. 96, pp. 1486–1491, February 1999 Genetics Global response of Saccharomyces cerevisiae to an alkylating agent SCOTT A. JELINSKY AND LEONA D. SAMSON* Department of Cancer Cell Biology, Division of Toxicology, Harvard School of Public Health, 665 Huntington Avenue, Boston, MA 02115 Edited by David Botstein, Stanford University School of Medicine, Stanford, CA, and approved December 7, 1998 (received for review September 16, 1998) ABSTRACT DNA chip technology enables simultaneous CUPr) was used in this study and was grown in 1% yeast examination of how '6,200 Saccharomyces cerevisiae gene tran- extracty2% peptoney2% glucose at 30°C. Cells were grown to a script levels, representing the entire genome, respond to envi- density of 5 3 106 cells per ml as measured by counting duplicated ronmental change. By using chips bearing oligonucleotide ar- dilutions. Cultures were split into two; MMS (0.1%) was added rays, we show that, after exposure to the alkylating agent methyl directly to one culture, and both cultures were incubated at 30°C ' methanesulfonate, 325 gene transcript levels are increased and for 1 h. Cells were pelleted and washed once in distilled H2O and '76 are decreased. Of the 21 genes that already were known to once in AE buffer (50 mM NaOAc, pH 5.2y10 mM EDTA) be induced by a DNA-damaging agent, 18 can be scored as immediately before RNA extraction. inducible in this data set, and surprisingly, most of the newly RNA Extraction. Total RNA was isolated by using a hot-phenol identified inducible genes are induced even more strongly than method (15). -

Supplementary Figure 1. Rpod Sequence Alignment. Protein Sequence Alignment Was Performed for Rpod from Three Species, Pseudoruegeria Sp
J. Microbiol. Biotechnol. https://doi.org/10.4014/jmb.1911.11006 J. Microbiol. Biotechnol. https://doi.org/10.4014/jmb.2003.03025 Supplementary Figure 1. RpoD sequence alignment. Protein sequence alignment was performed for RpoD from three species, Pseudoruegeria sp. M32A2M (FPS10_24745, this study), Ruegeria pomeroyi (NCBI RefSeq: WP_011047484.1), and Escherichia coli (NCBI RefSeq: NP_417539.1). Multalin version 5.4.1 was used for the analysis. Subregion 2 and 4 are represented in a rectangle, and the helix-turn-helix motif in subregion 4 is highlighted in red. The amino acid degeneration from any of the three was highlighted in light gray and the sequence degeneration between Pseudoruegeria sp. M32A2M and R. pomeroyi is highlighted in dark gray. The substituted two amino acids into HTH motif (K578Q and D581S) against E. coli were marked as asterisk. Supplementary Table 1. Genome assembly statistics Categories Pseudoruegeria sp. M32A2M Number of scaffolds less than 1,000 bp 0 Number of scaffolds between 1,000 bp –10,000 bp 39 Number of scaffolds between 10,000 bp – 100,000 bp 38 Number of scaffolds larger than 100,000 bp 14 Number of scaffolds 91 Total assembled length (bp) 5,466,515 G+C contents (%) 62.4 N50 (bp) 249,384 Minimum length of scaffold (bp) 1,015 Maximum length of scaffold (bp) 733,566 Total Ns included in the draft genome 2,158 Supplementary Table 2. The list of gene annotation and its functional categorization in Pseudoruegeria sp. -
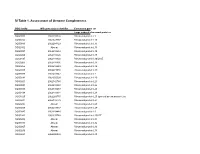
SI Table 1. Assessment of Genome Completeness
SI Table 1. Assessment of Genome Completeness COG family IMG gene object identifier Conserved gene set Large subunit ribosomal proteins COG0081 2062288324 Ribosomal protein L1 COG0244 2062347387 Ribosomal protein L10 COG0080 2062288323 Ribosomal protein L11 COG0102 Absent Ribosomal protein L13 COG0093 2062418832 Ribosomal protein L14 COG0200 2062418826 Ribosomal protein L15 COG0197 2062418838 Ribosomal protein L16/L10E COG0203 2062418836 Ribosomal protein L17 COG0256 2062418829 Ribosomal protein L18 COG0335 2062273558 Ribosomal protein L19 COG0090 2062418842 Ribosomal protein L2 COG0292 2062350539 Ribosomal protein L20 COG0261 2062142780 Ribosomal protein L21 COG0091 2062418840 Ribosomal protein L22 COG0089 2062138283 Ribosomal protein L23 COG0198 2062418834 Ribosomal protein L24 COG1825 2062269715 Ribosomal protein L25 (general stress protein Ctc) COG0211 2062142779 Ribosomal protein L27 COG0227 Absent Ribosomal protein L28 COG0255 2062418837 Ribosomal protein L29 COG0087 2062154483 Ribosomal protein L3 COG1841 2062335748 Ribosomal protein L30/L7E COG0254 Absent Ribosomal protein L31 COG0333 Absent Ribosomal protein L32 COG0267 Absent Ribosomal protein L33 COG0230 Absent Ribosomal protein L34 COG0291 2062350538 Ribosomal protein L35 COG0257 Absent Ribosomal protein L36 COG0088 2062138282 Ribosomal protein L4 COG0094 2062418833 Ribosomal protein L5 COG0097 2062418830 Ribosomal protein L6P/L9E COG0222 2062288326 Ribosomal protein L7/L12 COG0359 2062209880 Ribosomal protein L9 Small subunit ribosomal proteins COG0539 Absent Ribosomal protein