Data-Driven Insights Into Ligands, Proteins, and Genetic Mutations
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Gentaur Products List
Chapter 2 : Gentaur Products List • Rabbit Anti LAMR1 Polyclonal Antibody Cy5 Conjugated Conjugated • Rabbit Anti Podoplanin gp36 Polyclonal Antibody Cy5 • Rabbit Anti LAMR1 CT Polyclonal Antibody Cy5 • Rabbit Anti phospho NFKB p65 Ser536 Polyclonal Conjugated Conjugated Antibody Cy5 Conjugated • Rabbit Anti CHRNA7 Polyclonal Antibody Cy5 Conjugated • Rat Anti IAA Monoclonal Antibody Cy5 Conjugated • Rabbit Anti EV71 VP1 CT Polyclonal Antibody Cy5 • Rabbit Anti Connexin 40 Polyclonal Antibody Cy5 • Rabbit Anti IAA Indole 3 Acetic Acid Polyclonal Antibody Conjugated Conjugated Cy5 Conjugated • Rabbit Anti LHR CGR Polyclonal Antibody Cy5 Conjugated • Rabbit Anti Integrin beta 7 Polyclonal Antibody Cy5 • Rabbit Anti Natrexone Polyclonal Antibody Cy5 Conjugated • Rabbit Anti MMP 20 Polyclonal Antibody Cy5 Conjugated Conjugated • Rabbit Anti Melamine Polyclonal Antibody Cy5 Conjugated • Rabbit Anti BCHE NT Polyclonal Antibody Cy5 Conjugated • Rabbit Anti NAP1 NAP1L1 Polyclonal Antibody Cy5 • Rabbit Anti Acetyl p53 K382 Polyclonal Antibody Cy5 • Rabbit Anti BCHE CT Polyclonal Antibody Cy5 Conjugated Conjugated Conjugated • Rabbit Anti HPV16 E6 Polyclonal Antibody Cy5 Conjugated • Rabbit Anti CCP Polyclonal Antibody Cy5 Conjugated • Rabbit Anti JAK2 Polyclonal Antibody Cy5 Conjugated • Rabbit Anti HPV18 E6 Polyclonal Antibody Cy5 Conjugated • Rabbit Anti HDC Polyclonal Antibody Cy5 Conjugated • Rabbit Anti Microsporidia protien Polyclonal Antibody Cy5 • Rabbit Anti HPV16 E7 Polyclonal Antibody Cy5 Conjugated • Rabbit Anti Neurocan Polyclonal -

Contig Protein Description Symbol Anterior Posterior Ratio
Table S2. List of proteins detected in anterior and posterior intestine pooled samples. Data on protein expression are mean ± SEM of 4 pools fed the experimental diets. The number of the contig in the Sea Bream Database (http://nutrigroup-iats.org/seabreamdb) is indicated. Contig Protein Description Symbol Anterior Posterior Ratio Ant/Pos C2_6629 1,4-alpha-glucan-branching enzyme GBE1 0.88±0.1 0.91±0.03 0.98 C2_4764 116 kDa U5 small nuclear ribonucleoprotein component EFTUD2 0.74±0.09 0.71±0.05 1.03 C2_299 14-3-3 protein beta/alpha-1 YWHAB 1.45±0.23 2.18±0.09 0.67 C2_268 14-3-3 protein epsilon YWHAE 1.28±0.2 2.01±0.13 0.63 C2_2474 14-3-3 protein gamma-1 YWHAG 1.8±0.41 2.72±0.09 0.66 C2_1017 14-3-3 protein zeta YWHAZ 1.33±0.14 4.41±0.38 0.30 C2_34474 14-3-3-like protein 2 YWHAQ 1.3±0.11 1.85±0.13 0.70 C2_4902 17-beta-hydroxysteroid dehydrogenase 14 HSD17B14 0.93±0.05 2.33±0.09 0.40 C2_3100 1-acylglycerol-3-phosphate O-acyltransferase ABHD5 ABHD5 0.85±0.07 0.78±0.13 1.10 C2_15440 1-phosphatidylinositol phosphodiesterase PLCD1 0.65±0.12 0.4±0.06 1.65 C2_12986 1-phosphatidylinositol-4,5-bisphosphate phosphodiesterase delta-1 PLCD1 0.76±0.08 1.15±0.16 0.66 C2_4412 1-phosphatidylinositol-4,5-bisphosphate phosphodiesterase gamma-2 PLCG2 1.13±0.08 2.08±0.27 0.54 C2_3170 2,4-dienoyl-CoA reductase, mitochondrial DECR1 1.16±0.1 0.83±0.03 1.39 C2_1520 26S protease regulatory subunit 10B PSMC6 1.37±0.21 1.43±0.04 0.96 C2_4264 26S protease regulatory subunit 4 PSMC1 1.2±0.2 1.78±0.08 0.68 C2_1666 26S protease regulatory subunit 6A PSMC3 1.44±0.24 1.61±0.08 -
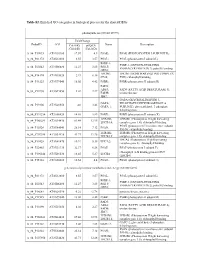
Table S2. Enriched GO Categories in Biological Process for the Shared Degs
Table S2. Enriched GO categories in biological process for the shared DEGs photosynthesis (GO ID:15979) Fold Change ProbeID AGI Col-0(R) pifQ(D) Name Description /Col-0(D) /Col-0(D) A_84_P19035 AT1G30380 17.07 4.9 PSAK; PSAK (PHOTOSYSTEM I SUBUNIT K) A_84_P21372 AT4G12800 8.55 3.57 PSAL; PSAL (photosystem I subunit L) PSBP-1; PSBP-1 (OXYGEN-EVOLVING A_84_P20343 AT1G06680 12.27 3.85 PSII-P; ENHANCER PROTEIN 2); poly(U) binding OEE2; LHCB6; LHCB6 (LIGHT HARVESTING COMPLEX A_84_P14174 AT1G15820 23.9 6.16 CP24; PSII); chlorophyll binding A_84_P11525 AT1G79040 16.02 4.42 PSBR; PSBR (photosystem II subunit R) FAD5; ADS3; FAD5 (FATTY ACID DESATURASE 5); A_84_P19290 AT3G15850 4.02 2.27 FADB; oxidoreductase JB67; GAPA (GLYCERALDEHYDE 3- GAPA; PHOSPHATE DEHYDROGENASE A A_84_P19306 AT3G26650 4.6 3.43 GAPA-1; SUBUNIT); glyceraldehyde-3-phosphate dehydrogenase A_84_P193234 AT2G06520 14.01 3.89 PSBX; PSBX (photosystem II subunit X) LHB1B1; LHB1B1 (Photosystem II light harvesting A_84_P160283 AT2G34430 89.44 32.95 LHCB1.4; complex gene 1.4); chlorophyll binding PSAN (photosystem I reaction center subunit A_84_P10324 AT5G64040 26.14 7.12 PSAN; PSI-N); calmodulin binding LHB1B2; LHB1B2 (Photosystem II light harvesting A_84_P207958 AT2G34420 41.71 12.26 LHCB1.5; complex gene 1.5); chlorophyll binding LHCA2 (Photosystem I light harvesting A_84_P19428 AT3G61470 10.91 5.36 LHCA2; complex gene 2); chlorophyll binding A_84_P22465 AT1G31330 32.37 6.58 PSAF; PSAF (photosystem I subunit F) chlorophyll A-B binding protein CP29 A_84_P190244 AT5G01530 16.45 5.27 LHCB4 -

Fatty Acid Biosynthesis
BI/CH 422/622 ANABOLISM OUTLINE: Photosynthesis Carbon Assimilation – Calvin Cycle Carbohydrate Biosynthesis in Animals Gluconeogenesis Glycogen Synthesis Pentose-Phosphate Pathway Regulation of Carbohydrate Metabolism Anaplerotic reactions Biosynthesis of Fatty Acids and Lipids Fatty Acids contrasts Diversification of fatty acids location & transport Eicosanoids Synthesis Prostaglandins and Thromboxane acetyl-CoA carboxylase Triacylglycerides fatty acid synthase ACP priming Membrane lipids 4 steps Glycerophospholipids Control of fatty acid metabolism Sphingolipids Isoprene lipids: Cholesterol ANABOLISM II: Biosynthesis of Fatty Acids & Lipids 1 ANABOLISM II: Biosynthesis of Fatty Acids & Lipids 1. Biosynthesis of fatty acids 2. Regulation of fatty acid degradation and synthesis 3. Assembly of fatty acids into triacylglycerol and phospholipids 4. Metabolism of isoprenes a. Ketone bodies and Isoprene biosynthesis b. Isoprene polymerization i. Cholesterol ii. Steroids & other molecules iii. Regulation iv. Role of cholesterol in human disease ANABOLISM II: Biosynthesis of Fatty Acids & Lipids Lipid Fat Biosynthesis Catabolism Fatty Acid Fatty Acid Degradation Synthesis Ketone body Isoprene Utilization Biosynthesis 2 Catabolism Fatty Acid Biosynthesis Anabolism • Contrast with Sugars – Lipids have have hydro-carbons not carbo-hydrates – more reduced=more energy – Long-term storage vs short-term storage – Lipids are essential for structure in ALL organisms: membrane phospholipids • Catabolism of fatty acids –produces acetyl-CoA –produces reducing -

Phylogenetic Analysis, Subcellular Localization, and Expression
BMC Plant Biology BioMed Central Research article Open Access Phylogenetic analysis, subcellular localization, and expression patterns of RPD3/HDA1 family histone deacetylases in plants Malona V Alinsug, Chun-Wei Yu and Keqiang Wu* Address: Institute of Plant Biology, College of Life Science, National Taiwan University, Taipei, Taiwan Email: Malona V Alinsug - [email protected]; Chun-Wei Yu - [email protected]; Keqiang Wu* - [email protected] * Corresponding author Published: 28 March 2009 Received: 26 November 2008 Accepted: 28 March 2009 BMC Plant Biology 2009, 9:37 doi:10.1186/1471-2229-9-37 This article is available from: http://www.biomedcentral.com/1471-2229/9/37 © 2009 Alinsug et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: Although histone deacetylases from model organisms have been previously identified, there is no clear basis for the classification of histone deacetylases under the RPD3/ HDA1 superfamily, particularly on plants. Thus, this study aims to reconstruct a phylogenetic tree to determine evolutionary relationships between RPD3/HDA1 histone deacetylases from six different plants representing dicots with Arabidopsis thaliana, Populus trichocarpa, and Pinus taeda, monocots with Oryza sativa and Zea mays, and the lower plants with Physcomitrella patens. Results: Sixty two histone deacetylases of RPD3/HDA1 family from the six plant species were phylogenetically analyzed to determine corresponding orthologues. Three clusters were formed separating Class I, Class II, and Class IV. -

Saturated Long-Chain Fatty Acid-Producing Bacteria Contribute
Zhao et al. Microbiome (2018) 6:107 https://doi.org/10.1186/s40168-018-0492-6 RESEARCH Open Access Saturated long-chain fatty acid-producing bacteria contribute to enhanced colonic motility in rats Ling Zhao1†, Yufen Huang2†, Lin Lu1†, Wei Yang1, Tao Huang1, Zesi Lin3, Chengyuan Lin1,4, Hiuyee Kwan1, Hoi Leong Xavier Wong1, Yang Chen5, Silong Sun2, Xuefeng Xie2, Xiaodong Fang2,5, Huanming Yang6, Jian Wang6, Lixin Zhu7* and Zhaoxiang Bian1* Abstract Background: The gut microbiota is closely associated with gastrointestinal (GI) motility disorder, but the mechanism(s) by which bacteria interact with and affect host GI motility remains unclear. In this study, through using metabolomic and metagenomic analyses, an animal model of neonatal maternal separation (NMS) characterized by accelerated colonic motility and gut dysbiosis was used to investigate the mechanism underlying microbiota-driven motility dysfunction. Results: An excess of intracolonic saturated long-chain fatty acids (SLCFAs) was associated with enhanced bowel motility in NMS rats. Heptadecanoic acid (C17:0) and stearic acid (C18:0), as the most abundant odd- and even- numbered carbon SLCFAs in the colon lumen, can promote rat colonic muscle contraction and increase stool frequency. Increase of SLCFAs was positively correlated with elevated abundances of Prevotella, Lactobacillus, and Alistipes. Functional annotation found that the level of bacterial LCFA biosynthesis was highly enriched in NMS group. Essential synthetic genes Fabs were largely identified from the genera Prevotella, Lactobacillus, and Alistipes. Pseudo germ-free (GF) rats receiving fecal microbiota from NMS donors exhibited increased defecation frequency and upregulated bacterial production of intracolonic SLCFAs. Modulation of gut dysbiosis by neomycin effectively attenuated GI motility and reduced bacterial SLCFA generation in the colon lumen of NMS rats. -

Crystal Structure of Fabz-ACP Complex Reveals a Dynamic Seesaw-Like Catalytic Mechanism of Dehydratase in Fatty Acid Biosynthesis
Cell Research (2016) 26:1330-1344. ORIGINAL ARTICLE www.nature.com/cr Crystal structure of FabZ-ACP complex reveals a dynamic seesaw-like catalytic mechanism of dehydratase in fatty acid biosynthesis Lin Zhang1, 2, Jianfeng Xiao3, Jianrong Xu1, Tianran Fu1, Zhiwei Cao1, Liang Zhu1, 2, Hong-Zhuan Chen1, 2, Xu Shen3, Hualiang Jiang3, Liang Zhang1, 2 1Department of Pharmacology and Chemical Biology, Shanghai Jiao Tong University School of Medicine, Shanghai, China; 2Shanghai Universities Collaborative Innovation Center for Translational Medicine, Shanghai, China; 3State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China Fatty acid biosynthesis (FAS) is a vital process in cells. Fatty acids are essential for cell assembly and cellular me- tabolism. Abnormal FAS directly correlates with cell growth delay and human diseases, such as metabolic syndromes and various cancers. The FAS system utilizes an acyl carrier protein (ACP) as a transporter to stabilize and shuttle the growing fatty acid chain throughout enzymatic modules for stepwise catalysis. Studying the interactions between enzymatic modules and ACP is, therefore, critical for understanding the biological function of the FAS system. How- ever, the information remains unclear due to the high flexibility of ACP and its weak interaction with enzymatic mod- ules. We present here a 2.55 Å crystal structure of type II FAS dehydratase FabZ in complex with holo-ACP, which exhibits a highly symmetrical FabZ hexamer-ACP3 stoichiometry with each ACP binding to a FabZ dimer subunit. Further structural analysis, together with biophysical and computational results, reveals a novel dynamic seesaw-like ACP binding and catalysis mechanism for the dehydratase module in the FAS system, which is regulated by a critical gatekeeper residue (Tyr100 in FabZ) that manipulates the movements of the β-sheet layer. -

The Structural Basis of Gas-Responsive Transcription by the Human Nuclear Hormone Receptor REV-Erbb
PLoS BIOLOGY The Structural Basis of Gas-Responsive Transcription by the Human Nuclear Hormone Receptor REV-ERBb Keith I. Pardee1,2, Xiaohui Xu1,2,3, Jeff Reinking1,2,7¤a, Anja Schuetz4¤b, Aiping Dong4, Suya Liu5, Rongguang Zhang6, Jens Tiefenbach1,2, Gilles Lajoie5, Alexander N. Plotnikov4¤b, Alexey Botchkarev4, Henry M. Krause1,2*, Aled Edwards1,2,3,4* 1 Banting and Best Department of Medical Research, The Department of Molecular Genetics, University of Toronto, Toronto, Canada, 2 Terrence Donnelly Centre for Cellular & Biomolecular Research, University of Toronto, Toronto, Canada, 3 Midwest Center for Structural Genomics, University of Toronto, Toronto, Canada, 4 Structural Genomics Consortium, University of Toronto, Toronto, Canada, 5 Department of Biochemistry, University of Western Ontario, London, Ontario, Canada, 6 Midwest Center for Structural Genomics, Argonne National Lab, Argonne, Illinois, United States of America, 7 Department of Biology, State University of New York at New Paltz, New Paltz, New York, United States of America Heme is a ligand for the human nuclear receptors (NR) REV-ERBa and REV-ERBb, which are transcriptional repressors that play important roles in circadian rhythm, lipid and glucose metabolism, and diseases such as diabetes, atherosclerosis, inflammation, and cancer. Here we show that transcription repression mediated by heme-bound REV- ERBs is reversed by the addition of nitric oxide (NO), and that the heme and NO effects are mediated by the C-terminal ligand-binding domain (LBD). A 1.9 A˚ crystal structure of the REV-ERBb LBD, in complex with the oxidized Fe(III) form of heme, shows that heme binds in a prototypical NR ligand-binding pocket, where the heme iron is coordinately bound by histidine 568 and cysteine 384. -

Study of the Role of the Adaptor Protein Myd88 in the Iron-Sensing Pathway
Université de Montréal Study of the role of the adaptor protein MyD88 in the iron-sensing pathway and of the effect of curcumin in the development of anemia in a DSS-induced colitis mouse model By Macha Samba Mondonga Department of Microbiology and Immunology Faculty of Medicine Thesis presented to the Faculty of Graduate Studies in partial fulfillment of the Ph.D. degree in Microbiology, Immunology and Infectious Diseases 30 Aout 2018 © Macha Samba Mondonga, 2018 Université de Montréal Cette thése est intitulée: Study of the role of the adaptor protein MyD88 in the iron-sensing pathway and of the effect of curcumin in the development of anemia in a DSS-induced colitis mouse model Par Macha Samba Mondonga Department of Microbiology and Immunology Faculty of Medicine 30 Aout 2018 © Macha Samba Mondonga, 2018 RÉSUMÉ Étude du rôle de la protéine adaptatrice MyD88 dans la voie de détection du fer et de l’effet de la curcumine dans le développement de l'anémie dans un modèle murin de colite induite par le DSS L'hepcidine est l'hormone peptidique essentielle pour la régulation systémique de l'homéostasie du fer, en déclenchant la dégradation de la ferroportine et en évitant le développement d'une surcharge ou d'une carence en fer. L'expression de l'ARNm de l'hepcidine HAMP est contrôlée par deux voies principales, la voix de signalisation inflammatoire et la voie de détection en fer, BMP/SMAD4. Il a été rapporté que MyD88, la protéine adaptatrice impliquée dans l'activation des cytokines via la stimulation des récepteurs toll-like (TLRs), contribue à l'expression de l'hepcidine dans la voie inflammatoire. -

Lysine Acetylation: Elucidating the Components of an Emerging Global Signaling Pathway in Trypanosomes
Hindawi Publishing Corporation Journal of Biomedicine and Biotechnology Volume 2012, Article ID 452934, 16 pages doi:10.1155/2012/452934 Review Article Lysine Acetylation: Elucidating the Components of an Emerging Global Signaling Pathway in Trypanosomes Victoria Lucia Alonso1, 2 and Esteban Carlos Serra1, 2 1 Departamento de Microbiolog´ıa, Facultad de Ciencias Bioqu´ımicas y Farmac´euticas, Universidad Nacional de Rosario, Suipacha 531, Rosario 2000, Argentina 2 Instituto de Biolog´ıa Molecular y Celular de Rosario, CONICET-UNR, Suipacha 590, Rosario 2000, Argentina Correspondence should be addressed to Esteban Carlos Serra, [email protected] Received 17 April 2012; Revised 20 July 2012; Accepted 30 July 2012 Academic Editor: Andrea Silvana Ropolo´ Copyright © 2012 V. L. Alonso and E. C. Serra. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. In the past ten years the number of acetylated proteins reported in literature grew exponentially. Several authors have proposed that acetylation might be a key component in most eukaryotic signaling pathways, as important as phosphorylation. The enzymes involved in this process are starting to emerge; acetyltransferases and deacetylases are found inside and outside the nuclear compartment and have different regulatory functions. In trypanosomatids several of these enzymes have been described and are postulated to be novel antiparasitic targets for the rational design of drugs. In this paper we overview the most important known acetylated proteins and the advances made in the identification of new acetylated proteins using high-resolution mass spectrometry. -

Enhanced Calcium Carbonate-Biofilm Complex Formation by Alkali
Lee and Park AMB Expr (2019) 9:49 https://doi.org/10.1186/s13568-019-0773-x ORIGINAL ARTICLE Open Access Enhanced calcium carbonate-bioflm complex formation by alkali-generating Lysinibacillus boronitolerans YS11 and alkaliphilic Bacillus sp. AK13 Yun Suk Lee and Woojun Park* Abstract Microbially induced calcium carbonate (CaCO3) precipitation (MICP) is a process where microbes induce condition favorable for CaCO3 formation through metabolic activities by increasing the pH or carbonate ions when calcium is near. The molecular and ecological basis of CaCO3 precipitating (CCP) bacteria has been poorly illuminated. Here, we showed that increased pH levels by deamination of amino acids is a driving force toward MICP using alkalitoler- ant Lysinibacillus boronitolerans YS11 as a model species of non-ureolytic CCP bacteria. This alkaline generation also facilitates the growth of neighboring alkaliphilic Bacillus sp. AK13, which could alter characteristics of MICP by chang- ing the size and shape of CaCO3 minerals. Furthermore, we showed CaCO3 that precipitates earlier in an experiment modifes membrane rigidity of YS11 strain via upregulation of branched chain fatty acid synthesis. This work closely examines MICP conditions by deamination and the efect of MICP on cell membrane rigidity and crystal formation for the frst time. Keywords: Alkaline generation, Dual species CaCO3 precipitation, Bacteria-CaCO3 interaction, Branched chain fatty acid synthesis, Membrane rigidity Introduction exopolysaccharide (EPS) formation, eventually leading to Calcium carbonate precipitating (CCP) bacteria con- microbially induced CaCO3 precipitation (MICP). Bac- tribute to the geochemical cycle as they precipitate car- terial metabolic pathways can create compounds that bonate minerals, including calcium carbonate in nature increase the solution pH. -

(GSNOR1) Function Leads to an Altered DNA and Histone Methylation Pattern in Arabidopsis Thaliana
TECHNISCHE UNIVERSITÄT MÜNCHEN Wissenschaftszentrum Weihenstephan für Ernährung, Landnutzung und Umwelt (WZW) Lehrstuhl für Biochemische Pflanzenpathologie Loss of S-NITROSOGLUTATHIONE REDUCTASE 1 (GSNOR1) function leads to an altered DNA and histone methylation pattern in Arabidopsis thaliana Eva Rudolf Vollständiger Abdruck der von der Fakultät Wissenschaftszentrum Weihenstephan für Ernährung, Landnutzung und Umwelt der Technischen Universität München zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften genehmigten Dissertation. Vorsitzender: Prof. Dr. Frank Johannes Prüfer der Dissertation: 1. Prof. Dr. Jörg Durner 2. apl. Prof. Dr. Ramon A. Torres Ruiz Die Dissertation wurde am 30.01.2020 bei der Technischen Universität München eingereicht und durch die Fakultät Wissenschaftszentrum Weihenstephan für Ernährung, Landnutzung und Umwelt am 20.04.2020 angenommen. To my family, Florian and Tobias. Publications and conference contributions related to this thesis: Izabella Kovacs, Alexandra Ageeva, Eva König and Christian Lindermayr, 2016. Chapter Two – S-Nitrosylation of Nuclear Proteins: New Pathways in Regulation of Gene Expression. In Advances in Botanical Research edited by David Wendehenne. Nitric Oxide and Signaling in Plants. Academic Press, 77, 15–39. Eva Rudolf, Markus Wirtz, Ignasi Forné and Christian Lindermayr. S-Nitrosothiols as architect of the methylome in Arabidopsis thaliana. EMBO Conference - Chromatin and Epigenetics 2017, Heidelberg, Germany, Poster. Eva Rudolf, Alexandra Ageeva-Kieferle, Alexander Mengel, Ignasi Forné, Rüdiger Hell, Axel Imhof, Markus Wirtz, Jörg Durner and Christian Lindermayr. Post-translational modification of histones: Nitric oxide modulates chromatin structure. Symposium - From Proteome to Phenotype: role of post- translational modifications 2017, Edinburgh, United Kingdom, Oral presentation. Alexandra Ageeva-Kieferle, Eva Rudolf and Christian Lindermayr, 2019. Redox-Dependent Chromatin Remodeling: A New Function of Nitric Oxide as Architect of Chromatin Structure in Plants.