Introduction: Schedule G List of Drugs Under Schedule G
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Review on the Current Classification and Regulatory Provisions for Medicines in Drug & Cosmetic Act, in the Light of Present Day Context
Section Pharmaindustry Commentary A Review on the Current Classification and Regulatory Provisions for Medicines in Drug & Cosmetic Act, in the light of Present Day Context Prashant Tandon1, Varun Gupta2, Ashish Ranjan3, Purav Gandhi4, Anand Kotiyal5, 3 Aastha Kapoor 3 1Founder ;2VP & Head Medical Affair; Manager Medical Affair; 5Drug Data Analyst Medical Affair, 1mg Technologies Private Limited, 4th Floor, Motorola Building, MG Road, Sector 14, Gurugram, Haryana, 122001. 4Founder, Remedy Social, C/602, Tulip Citadel, Shreyas Tekra, Ambawadi, Ahmedabad 380015, Gujarat. ABSTRACT______________________________________________________________ Background: Current classification of medicines in Conclusions: We have recommended a revised drug India under Drug and Cosmetic Act into Schedule G, classification system that is more comprehensive in coverage and H, H1, X is outdated, evolved through patchwork over eliminates the overlaps between classes. Moreover, considering the years and needs to be thoroughly updated. The the implementation challenges for such a drug classification primary aim of the scheduling system is to ensure system in the diverse and fragmented ecosystem in India, we appropriate access to medicines while balancing recommend a technology backed platform to help monitor the public health and safety. India is experiencing a rapid implementation. transition with the rising burden of chronic non- communicable diseases where regular access of Key words: Drug Classification System, Drug and Cosmetic Act affordable medicines is critical for chronic disease India, Digitization of Prescriptions, Drug Schedules in India, management to prevent complications. Methods: We Schedule H, Monitoring Drug Schedule System analyzed drugs commonly selling across India, Received: 01.09.17 | Accepted:16.09.17 through multiple information sources including 1mg drug database, PharmaTrac (AIOCD-AWACS), Corresponding Author inventory data from distributors and retailers, Dr. -

(12) Patent Application Publication (10) Pub. No.: US 2010/0221245 A1 Kunin (43) Pub
US 2010O221245A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2010/0221245 A1 Kunin (43) Pub. Date: Sep. 2, 2010 (54) TOPICAL SKIN CARE COMPOSITION Publication Classification (51) Int. Cl. (76) Inventor: Audrey Kunin, Mission Hills, KS A 6LX 39/395 (2006.01) (US) A6II 3L/235 (2006.01) A638/16 (2006.01) Correspondence Address: (52) U.S. Cl. ......................... 424/133.1: 514/533: 514/12 HUSCH BLACKWELL SANDERS LLP (57) ABSTRACT 4801 Main Street, Suite 1000 - KANSAS CITY, MO 64112 (US) The present invention is directed to a topical skin care com position. The composition has the unique ability to treat acne without drying out the user's skin. In particular, the compo (21) Appl. No.: 12/395,251 sition includes a base, an antibacterial agent, at least one anti-inflammatory agent, and at least one antioxidant. The (22) Filed: Feb. 27, 2009 antibacterial agent may be benzoyl peroxide. US 2010/0221 245 A1 Sep. 2, 2010 TOPCAL SKIN CARE COMPOSITION stay of acne treatment since the 1950s. Skin irritation is the most common side effect of benzoyl peroxide and other anti BACKGROUND OF THE INVENTION biotic usage. Some treatments can be severe and can leave the 0001. The present invention generally relates to composi user's skin excessively dry. Excessive use of some acne prod tions and methods for producing topical skin care. Acne Vul ucts may cause redness, dryness of the face, and can actually garis, or acne, is a common skin disease that is prevalent in lead to more acne. Therefore, it would be beneficial to provide teenagers and young adults. -

Wo 2010/075090 A2
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date 1 July 2010 (01.07.2010) WO 2010/075090 A2 (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every C07D 409/14 (2006.01) A61K 31/7028 (2006.01) kind of national protection available): AE, AG, AL, AM, C07D 409/12 (2006.01) A61P 11/06 (2006.01) AO, AT, AU, AZ, BA, BB, BG, BH, BR, BW, BY, BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, (21) International Application Number: DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, PCT/US2009/068073 HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KM, KN, KP, (22) International Filing Date: KR, KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, 15 December 2009 (15.12.2009) ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, PE, PG, PH, PL, PT, RO, RS, RU, SC, SD, (25) Filing Language: English SE, SG, SK, SL, SM, ST, SV, SY, TJ, TM, TN, TR, TT, (26) Publication Language: English TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (30) Priority Data: (84) Designated States (unless otherwise indicated, for every 61/122,478 15 December 2008 (15.12.2008) US kind of regional protection available): ARIPO (BW, GH, GM, KE, LS, MW, MZ, NA, SD, SL, SZ, TZ, UG, ZM, (71) Applicant (for all designated States except US): AUS- ZW), Eurasian (AM, AZ, BY, KG, KZ, MD, RU, TJ, PEX PHARMACEUTICALS, INC. -

Title 16. Crimes and Offenses Chapter 13. Controlled Substances Article 1
TITLE 16. CRIMES AND OFFENSES CHAPTER 13. CONTROLLED SUBSTANCES ARTICLE 1. GENERAL PROVISIONS § 16-13-1. Drug related objects (a) As used in this Code section, the term: (1) "Controlled substance" shall have the same meaning as defined in Article 2 of this chapter, relating to controlled substances. For the purposes of this Code section, the term "controlled substance" shall include marijuana as defined by paragraph (16) of Code Section 16-13-21. (2) "Dangerous drug" shall have the same meaning as defined in Article 3 of this chapter, relating to dangerous drugs. (3) "Drug related object" means any machine, instrument, tool, equipment, contrivance, or device which an average person would reasonably conclude is intended to be used for one or more of the following purposes: (A) To introduce into the human body any dangerous drug or controlled substance under circumstances in violation of the laws of this state; (B) To enhance the effect on the human body of any dangerous drug or controlled substance under circumstances in violation of the laws of this state; (C) To conceal any quantity of any dangerous drug or controlled substance under circumstances in violation of the laws of this state; or (D) To test the strength, effectiveness, or purity of any dangerous drug or controlled substance under circumstances in violation of the laws of this state. (4) "Knowingly" means having general knowledge that a machine, instrument, tool, item of equipment, contrivance, or device is a drug related object or having reasonable grounds to believe that any such object is or may, to an average person, appear to be a drug related object. -

Rhinitis - Allergic (1 of 15)
Rhinitis - Allergic (1 of 15) 1 Patient presents w/ signs & symptoms of rhinitis 2 • Consider other classifi cations of rhinitis DIAGNOSIS No - Please see Rhinitis Is allergic rhinitis - Nonallergic disease confi rmed? management chart Yes 3 ASSESS DURATION & SEVERITY OF ALLERGIC RHINITIS A Non-pharmacological therapy • Allergen avoidance • Patient education VAS <5 VAS ≥5 B Pharmacological therapy B Pharmacological therapy • Antihistamines (oral/nasal), &/or • Corticosteroids (nasal), w/ or without • Corticosteroids (nasal), or • Antihistamines (nasal), or • Cromone (nasal), or • LTRA • Leukotriene receptor antagonists (LTRA)MIMS TREATMENT © See next page Specifi cally for patients w/ asthma Not all products are available or approved for above use in all countries. Specifi c prescribing information may be found in the latest MIMS. B167 © MIMS Pediatrics 2020 Rhinitis - Allergic (2 of 15) Previously treated symptomatic Previously treated symptomatic patient (VAS <5) on antihistamines patient (VAS ≥5) on intensifi ed (oral/nasal) &/or corticosteroids (nasal) therapy w/ corticosteroids (nasal) w/ or without antihistamines (nasal) Intermittent Persistent symptoms, symptoms or without allergen w/ allergen exposure exposure B Pharmacological therapy B Pharmacological therapy • Step down or discontinue therapy • Continue or step up therapy Untreated REASSESS DISEASE SEVERITY VAS symptomatic patient DAILY UP TO DAY 3 (VAS <5 or ≥5) 4 CONTINUE Yes THERAPY & STEP EVALUATION VAS <5 DOWN THERAPY1 Improvement of symptoms? No VAS ≥5 B Pharmacological therapy • Step-up therapy REASSESS DISEASE SEVERITY MIMSVAS DAILY UP TO DAY 7 4 Yes EVALUATION VAS <5 Improvement of symptoms? No TREATMENT © VAS ≥5 See next page Continue therapy if symptomatic; consider step-down or discontinuation of therapy if symptoms subside Not all products are available or approved for above use in all countries. -

(19) United States (12) Patent Application Publication (10) Pub
US 20130210835A1 (19) United States (12) Patent Application Publication (10) Pub. N0.2 US 2013/0210835 A1 Mitchell (43) Pub. Date: Aug. 15, 2013 (54) PHARMACEUTICAL COMPOSITIONS Publication Classi?cation (75) Inventor: Odes W. Mitchell; Arlington, TX (U S) (51) Int. Cl. A61K31/137 (2006.01) _ A611; 31/4402 (2006.01) (73) Ass1gnee: GM PHARMACEUTICAL, INC, A61K 31/485 (200601) Arhngton, TX (Us) A611; 31/09 (2006.01) _ A611; 31/495 (2006.01) (21) App1.No.. 13/703,584 A61K31/505 (200601) 22 PCT P1 d: J .13 2011 (52) us Cl ( ) 1e “n ’ CPC ........... .. A611; 31/137 (2013.01); A611;31/495 (86) PCT NO. PCT/“11,4031 (2013.01); A611;31/505 (2013.01); A611; 31/485 (2013.01); A611; 31/09 (2013.01); § 371 (0)0). A611;31/4402 (2013.01) (2), (4) Date: Feb- 2, 2013 USPC .... .. 514/255.04; 564/355; 514/653; 544/396; 544/332; 514/275; 546/74; 514/289; 514/282; Related US. Application Data 514657; 514652 (60) Provisional application No. 61/354,061; ?led on Jun. (57) ABSTRACT 11; 2010; provisional application No. 61/354,057; A composition of an antitussive; a decongestant; or an anti ?led on Jun. 11; 2010; provisional application No. histamine to treat respiratory and oral pharyngeal congestion 61/354,053; ?led on Jun. 11,2010. and related symptoms in a patient. US 2013/0210835 A1 Aug. 15,2013 PHARMACEUTICAL COMPOSITIONS mucus build-up to clear congestion in the air passages. Symp toms due to allergies or allergens are often treated With an CROSS-REFERENCES TO RELATED antihistamine. -

Withdrawing Drugs in the U.S. Versus Other Countries Benson Ninan
Volume 3 | Number 3 Article 87 2012 Withdrawing Drugs in the U.S. Versus Other Countries Benson Ninan Albert I. Wertheimer Follow this and additional works at: http://pubs.lib.umn.edu/innovations Recommended Citation Ninan B, Wertheimer AI. Withdrawing Drugs in the U.S. Versus Other Countries. Inov Pharm. 2012;3(3): Article 87. http://pubs.lib.umn.edu/innovations/vol3/iss3/6 INNOVATIONS in pharmacy is published by the University of Minnesota Libraries Publishing. Commentary POLICY Withdrawing Drugs in the U.S. Versus Other Countries Benson Ninan, Pharm.D.1 and Albert I Wertheimer, PhD, MBA2 1Pharmacy Intern, Rite Aid Pharmacies, Philadelphia, PA and 2Temple University School of Pharmacy, Philadelphia PA Key Words: Drug withdrawals, dangerous drugs, UN Banned Drug list Abstract Since 1979, the United Nations has maintained a list of drugs banned from sale in member countries. Interestingly, there are a number of pharmaceuticals on the market in the USA that have been banned elsewhere and similarly, there are some drug products that have been banned in the United States, but remain on the market in other countries. This report provides a look into the policies for banning drug sales internationally and the role of the United Nations in maintaining the master list for companies and countries to use for local decision guidance. Background recently updated issue is the fourteenth issue, which contains At present, one of the leading causes of death in the U.S. is data on 66 new products with updated/new information on believed to be adverse drug reactions.1-14 More than 20 22 existing products. -

Pharmaceutical Appendix to the Tariff Schedule 2
Harmonized Tariff Schedule of the United States (2007) (Rev. 2) Annotated for Statistical Reporting Purposes PHARMACEUTICAL APPENDIX TO THE HARMONIZED TARIFF SCHEDULE Harmonized Tariff Schedule of the United States (2007) (Rev. 2) Annotated for Statistical Reporting Purposes PHARMACEUTICAL APPENDIX TO THE TARIFF SCHEDULE 2 Table 1. This table enumerates products described by International Non-proprietary Names (INN) which shall be entered free of duty under general note 13 to the tariff schedule. The Chemical Abstracts Service (CAS) registry numbers also set forth in this table are included to assist in the identification of the products concerned. For purposes of the tariff schedule, any references to a product enumerated in this table includes such product by whatever name known. ABACAVIR 136470-78-5 ACIDUM LIDADRONICUM 63132-38-7 ABAFUNGIN 129639-79-8 ACIDUM SALCAPROZICUM 183990-46-7 ABAMECTIN 65195-55-3 ACIDUM SALCLOBUZICUM 387825-03-8 ABANOQUIL 90402-40-7 ACIFRAN 72420-38-3 ABAPERIDONUM 183849-43-6 ACIPIMOX 51037-30-0 ABARELIX 183552-38-7 ACITAZANOLAST 114607-46-4 ABATACEPTUM 332348-12-6 ACITEMATE 101197-99-3 ABCIXIMAB 143653-53-6 ACITRETIN 55079-83-9 ABECARNIL 111841-85-1 ACIVICIN 42228-92-2 ABETIMUSUM 167362-48-3 ACLANTATE 39633-62-0 ABIRATERONE 154229-19-3 ACLARUBICIN 57576-44-0 ABITESARTAN 137882-98-5 ACLATONIUM NAPADISILATE 55077-30-0 ABLUKAST 96566-25-5 ACODAZOLE 79152-85-5 ABRINEURINUM 178535-93-8 ACOLBIFENUM 182167-02-8 ABUNIDAZOLE 91017-58-2 ACONIAZIDE 13410-86-1 ACADESINE 2627-69-2 ACOTIAMIDUM 185106-16-5 ACAMPROSATE 77337-76-9 -

(19) 11 Patent Number: 6165500
USOO6165500A United States Patent (19) 11 Patent Number: 6,165,500 Cevc (45) Date of Patent: *Dec. 26, 2000 54 PREPARATION FOR THE APPLICATION OF WO 88/07362 10/1988 WIPO. AGENTS IN MINI-DROPLETS OTHER PUBLICATIONS 75 Inventor: Gregor Cevc, Heimstetten, Germany V.M. Knepp et al., “Controlled Drug Release from a Novel Liposomal Delivery System. II. Transdermal Delivery Char 73 Assignee: Idea AG, Munich, Germany acteristics” on Journal of Controlled Release 12(1990) Mar., No. 1, Amsterdam, NL, pp. 25–30. (Exhibit A). * Notice: This patent issued on a continued pros- C.E. Price, “A Review of the Factors Influencing the Pen ecution application filed under 37 CFR etration of Pesticides Through Plant Leaves” on I.C.I. Ltd., 1.53(d), and is subject to the twenty year Plant Protection Division, Jealott's Hill Research Station, patent term provisions of 35 U.S.C. Bracknell, Berkshire RG12 6EY, U.K., pp. 237-252. 154(a)(2). (Exhibit B). K. Karzel and R.K. Liedtke, “Mechanismen Transkutaner This patent is Subject to a terminal dis- Resorption” on Grandlagen/Basics, pp. 1487–1491. (Exhibit claimer. C). Michael Mezei, “Liposomes as a Skin Drug Delivery Sys 21 Appl. No.: 07/844,664 tem” 1985 Elsevier Science Publishers B.V. (Biomedical Division), pp 345-358. (Exhibit E). 22 Filed: Apr. 8, 1992 Adrienn Gesztes and Michael Mazei, “Topical Anesthesia of 30 Foreign Application Priority Data the Skin by Liposome-Encapsulated Tetracaine” on Anesth Analg 1988; 67: pp 1079–81. (Exhibit F). Aug. 24, 1990 DE) Germany ............................... 40 26834 Harish M. Patel, "Liposomes as a Controlled-Release Sys Aug. -
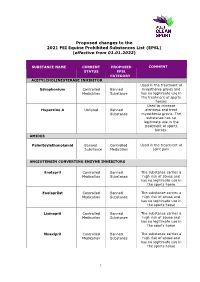
Proposed Changes to the 2021 FEI Equine Prohibited Substances List (EPSL) (Effective from 01.01.2022)
Proposed changes to the 2021 FEI Equine Prohibited Substances List (EPSL) (effective from 01.01.2022) SUBSTANCE NAME CURRENT PROPOSED COMMENT STATUS EPSL CATEGORY ACETYLCHOLINESTERASE INHIBITOR Used in the treatment of Edrophonium Controlled Banned myasthenia gravis and Medication Substance has no legitimate use in the treatment of sports horses Used to increase Huperzine A Unlisted Banned alertness and treat Substance myasthenia gravis. The substance has no legitimate use in the treatment of sports horses. AMIDES Palmitoylethanolamid Banned Controlled Used in the treatment of Substance Medication joint pain ANGIOTENSIN CONVERTING ENZYME INHIBITORS Enalapril Controlled Banned The substance carries a Medication Substance high risk of abuse and has no legitimate use in the sports horse Enalaprilat Controlled Banned The substance carries a Medication Substance high risk of abuse and has no legitimate use in the sports horse Lisinopril Controlled Banned The substance carries a Medication Substance high risk of abuse and has no legitimate use in the sports horse Moexipril Controlled Banned The substance carries a Medication Substance high risk of abuse and has no legitimate use in the sports horse 1 Perindoprilat Controlled Banned The substance carries a Medication Substance high risk of abuse and has no legitimate use in the sports horse ANTIHISTAMINES Antazoline Controlled Banned The substance has no Medication Substance legitimate use in the sports horse Azatadine Controlled Banned The substance has Medication Substance sedative effects -

Family Medicine (Fm-1) Multiple Choice Questions / Type I
FAMILY MEDICINE (FM-1) MULTIPLE CHOICE QUESTIONS / TYPE I Select the correct answers to the following questions!!! ...each question may have more than one correct answer. FM-1.1. (g) Multiple Choice Question Renal calcification is a possible complication of: A) medullary cystic kidney disease B) renal tuberculosis C) sarcoidosis D) sickle cell anemia E) secondary hyperparathyroidism FM-1.2. Multiple Choice Question CV, IM Which of the following statements concerning chromosomes are correct? A) their number is normally 46 B) mosaicism is the coexistence of cells with different number of chromosomes within the same organism C) they are always identical in cells of the same phenotype D) nondisjunction must be followed by translocation E) they can be used as tumor markers FM-1.3. Multiple Choice Question IM Drugs with a bacteriostatic effect in regular doses include: A) tetracyclines B) cephalosporins C) sulfamethoxazole and trimethoprim (Sumetrolim) D) erythromycin E) amoxycillin FM-1.4. (a Multiple Choice Question Factors causing a susceptibility to urinary tract infect include: A) urinary tract obstruction B) diabetes mellitus C) hyperkalemia D) prolonged tetracycline therapy E) pregnancy 2 Multiple Choice Questions / Type I • FAMILY MEDICINE (FM-1) FM-1.5. Multiple Choice Question ~&, IM Case Study: The medical history of a 45-year-old male reveals episodes of vertigo and loss of consciousness associated with sweating. Possible causes of his symptoms include: A) hyperventilation B) hyperglycemia C) Zollinger-Ellison syndrome D) pheochromocytoma E) paroxysmal tachycardia FM-1.6. Multiple Choice Question IM Possible causes of hematemesis include: A) salicylate administration B) an oral iron supplement overdose C) severe burn injury D) Menetrier's disease (giant hypertrophic gastritis) E) feeding via a nasogastric tube FM-1.7. -

BMJ Open Is Committed to Open Peer Review. As Part of This Commitment We Make the Peer Review History of Every Article We Publish Publicly Available
BMJ Open is committed to open peer review. As part of this commitment we make the peer review history of every article we publish publicly available. When an article is published we post the peer reviewers’ comments and the authors’ responses online. We also post the versions of the paper that were used during peer review. These are the versions that the peer review comments apply to. The versions of the paper that follow are the versions that were submitted during the peer review process. They are not the versions of record or the final published versions. They should not be cited or distributed as the published version of this manuscript. BMJ Open is an open access journal and the full, final, typeset and author-corrected version of record of the manuscript is available on our site with no access controls, subscription charges or pay-per-view fees (http://bmjopen.bmj.com). If you have any questions on BMJ Open’s open peer review process please email [email protected] BMJ Open Pediatric drug utilization in the Western Pacific region: Australia, Japan, South Korea, Hong Kong and Taiwan Journal: BMJ Open ManuscriptFor ID peerbmjopen-2019-032426 review only Article Type: Research Date Submitted by the 27-Jun-2019 Author: Complete List of Authors: Brauer, Ruth; University College London, Research Department of Practice and Policy, School of Pharmacy Wong, Ian; University College London, Research Department of Practice and Policy, School of Pharmacy; University of Hong Kong, Centre for Safe Medication Practice and Research, Department