Regioselectivity in the Cyclization of Delta, Epsilon-Epoxy Esters
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Extraction of Wax from Sorghum Bran •
i EXTRACTION OF WAX FROM SORGHUM BRAN • 1 HSIEN-WEN HSU <& . ***£/ B. S., National Taiwan University, China, 1951 A THESIS » submitted in partial fulfillment of the requirements for the degree MASTER OF SCIENCE - Department of Chemical Engineering KANSAS STATE COLLEGE OF AGRICULTURE AND APPLIED SCIENCE - 1955 » k Lo i~i ii Q^cu^v^Js TABLE OF CONTENTS INTRODUCTION 1 PREVIOUS WORK 3 MATERIALS 6 GLASS SOXHLET EXTRACTION AND ANALYSIS OF WAX AND OIL IN MISCELLA 7 Procedure of Using Soxhlet Extractor 7 Separation of Wax from Oil by the Acetone Method.. 8 Summary of Results 9 PILOT PLANT EXTRACTION 15 SEPARATION OF WAX AND OIL 16 Effect of Temperature in the Acetone Method 16 Urea Complex Method of Separation. 17 Theory 17 Experimental Procedure 22 Summary of Results 26 COST ESTIMATION OF A WAX RECOVERY PLANT 29 DISCUSSION 30 CONCLUSIONS 35 ACKNOWLEDGMENTS 36 BIBLIOGRAPHY 37 APPENDIX 39 INTRODUCTION i One of the functions of the Agricultural Experiment Station is the never ending search for means of better and fuller utilization of the agri- cultural products that are of particular interest in the state of Kansas. In line with this broad objective, the extraction of economically valuable lipid materials from the sorghum bran has been studied in several theses in the Departments of Chemical Engineering and Chemistry in recent years. Generally the emphasis has b. en placed upon either the chemical and physical properties of these lipides or the equipment design and performance for the solvent ex- traction operation. A brief review of the previous work is presented in the ensuing section. -

The Separation of Three Azeotropes by Extractive Distillation by An-I Yeh A
The separation of three azeotropes by extractive distillation by An-I Yeh A thesis submitted in partial fulfillment of the requirement for the degree of Master of Science in Chemical Engineering Montana State University © Copyright by An-I Yeh (1983) Abstract: Several different kinds of extractive distillation agents were investigated to affect the separation of three binary liquid mixtures, isopropyl ether - acetone, methyl acetate - methanol, and isopropyl ether - methyl ethyl ketone. Because of the small size of the extractive distillation column, relative volatilities were assumed constant and the Fenske equation was used to calculate the relative volatilities and the number of minimum theoretical plates. Dimethyl sulfoxide was found to be a good extractive distillation agent. Extractive distillation when employing a proper agent not only negated the azeotropes of the above mixtures, but also improved the efficiency of separation. This process could reverse the relative volatility of isopropyl ether and acetone. This reversion was also found in the system of methyl acetate and methanol when nitrobenzene was the agent. However, normal distillation curves were obtained for the system of isopropyl ether and methyl ethyl ketone undergoing extractive distillation. In the system of methyl acetate and methanol, the relative volatility decreased as the agents' carbon number increased when glycols were used as the agents. In addition, the oxygen number and the locations of hydroxyl groups in the glycols used were believed to affect the values of relative volatility. An appreciable amount of agent must be maintained in the column to affect separation. When dimethyl sulfoxide was an agent for the three systems studied, the relative volatility increased as the addition rate increased. -

Distillation 65 Chem 355 Jasperse DISTILLATION
Distillation 65 Chem 355 Jasperse DISTILLATION Background Distillation is a widely used technique for purifying liquids. The basic distillation process involves heating a liquid such that liquid molecules vaporize. The vapors produced are subsequently passed through a water-cooled condenser. Upon cooling, the vapor returns to it’s liquid phase. The liquid can then be collected. The ability to separate mixtures of liquids depends on differences in volatility (the ability to vaporize). For separation to occur, the vapor that is condensed and collected must be more pure than the original liquid mix. Distillation can be used to remove a volatile solvent from a nonvolatile product; to separate a volatile product from nonvolatile impurities; or to separate two or more volatile products that have sufficiently different boiling points. Vaporization and Boiling When a liquid is placed in a closed container, some of the molecules evaporate into any unoccupied space in the container. Evaporation, which occurs at temperatures below the boiling point of a compound, involves the transition from liquid to vapor of only those molecules at the liquid surface. Evaporation continues until an equilibrium is reached between molecules entering and leaving the liquid and vapor states. The pressure exerted by these gaseous molecules on the walls of the container is the equilibrium vapor pressure. The magnitude of this vapor pressure depends on the physical characteristics of the compound and increases as temperature increases. In an open container, equilibrium is never established, the vapor can simply leave, and the liquid eventually disappears. But whether in an open or closed situation, evaporation occurs only from the surface of the liquid. -
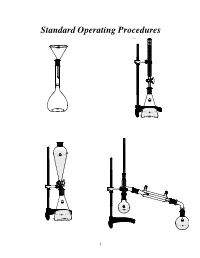
Standard Operating Procedures
Standard Operating Procedures 1 Standard Operating Procedures OVERVIEW In the following laboratory exercises you will be introduced to some of the glassware and tech- niques used by chemists to isolate components from natural or synthetic mixtures and to purify the individual compounds and characterize them by determining some of their physical proper- ties. While working collaboratively with your group members you will become acquainted with: a) Volumetric glassware b) Liquid-liquid extraction apparatus c) Distillation apparatus OBJECTIVES After finishing these sessions and reporting your results to your mentor, you should be able to: • Prepare solutions of exact concentrations • Separate liquid-liquid mixtures • Purify compounds by recrystallization • Separate mixtures by simple and fractional distillation 2 EXPERIMENT 1 Glassware Calibration, Primary and Secondary Standards, and Manual Titrations PART 1. Volumetric Glassware Calibration Volumetric glassware is used to either contain or deliver liquids at a specified temperature. Glassware manufacturers indicate this by inscribing on the volumetric ware the initials TC (to contain) or TD (to deliver) along with the calibration temperature, which is usually 20°C1. Volumetric glassware must be scrupulously clean before use. The presence of streaks or droplets is an indication of the presence of a grease film. To eliminate grease from glassware, scrub with detergent solution, rinse with tap water, and finally rinse with a small portion of distilled water. Volumetric flasks (TC) A volumetric flask has a large round bottom with only one graduation mark positioned on the long narrow neck. Graduation Mark Stopper The position of the mark facilitates the accurate and precise reading of the meniscus. If the flask is used to prepare a solution starting with a solid compound, add small amounts of sol- vent until the entire solid dissolves. -

Organic Chemistry
Wisebridge Learning Systems Organic Chemistry Reaction Mechanisms Pocket-Book WLS www.wisebridgelearning.com © 2006 J S Wetzel LEARNING STRATEGIES CONTENTS ● The key to building intuition is to develop the habit ALKANES of asking how each particular mechanism reflects Thermal Cracking - Pyrolysis . 1 general principles. Look for the concepts behind Combustion . 1 the chemistry to make organic chemistry more co- Free Radical Halogenation. 2 herent and rewarding. ALKENES Electrophilic Addition of HX to Alkenes . 3 ● Acid Catalyzed Hydration of Alkenes . 4 Exothermic reactions tend to follow pathways Electrophilic Addition of Halogens to Alkenes . 5 where like charges can separate or where un- Halohydrin Formation . 6 like charges can come together. When reading Free Radical Addition of HX to Alkenes . 7 organic chemistry mechanisms, keep the elec- Catalytic Hydrogenation of Alkenes. 8 tronegativities of the elements and their valence Oxidation of Alkenes to Vicinal Diols. 9 electron configurations always in your mind. Try Oxidative Cleavage of Alkenes . 10 to nterpret electron movement in terms of energy Ozonolysis of Alkenes . 10 Allylic Halogenation . 11 to make the reactions easier to understand and Oxymercuration-Demercuration . 13 remember. Hydroboration of Alkenes . 14 ALKYNES ● For MCAT preparation, pay special attention to Electrophilic Addition of HX to Alkynes . 15 Hydration of Alkynes. 15 reactions where the product hinges on regio- Free Radical Addition of HX to Alkynes . 16 and stereo-selectivity and reactions involving Electrophilic Halogenation of Alkynes. 16 resonant intermediates, which are special favor- Hydroboration of Alkynes . 17 ites of the test-writers. Catalytic Hydrogenation of Alkynes. 17 Reduction of Alkynes with Alkali Metal/Ammonia . 18 Formation and Use of Acetylide Anion Nucleophiles . -

United States Patent (10) Patent No.: US 9,586,922 B2 Wood Et Al
USOO9586922B2 (12) United States Patent (10) Patent No.: US 9,586,922 B2 Wood et al. (45) Date of Patent: Mar. 7, 2017 (54) METHODS FOR PURIFYING 9,388,150 B2 * 7/2016 Kim ..................... CO7D 307/48 5-(HALOMETHYL)FURFURAL 9,388,151 B2 7/2016 Browning et al. 2007/O161795 A1 7/2007 Cvak et al. 2009, 0234142 A1 9, 2009 Mascal (71) Applicant: MICROMIDAS, INC., West 2010, 0083565 A1 4, 2010 Gruter Sacramento, CA (US) 2010/0210745 A1 8/2010 McDaniel et al. 2011 O144359 A1 6, 2011 Heide et al. (72) Inventors: Alex B. Wood, Sacramento, CA (US); 2014/01 00378 A1 4/2014 Masuno et al. Shawn M. Browning, Sacramento, CA 2014/O1878O2 A1 7/2014 Mikochik et al. (US);US): Makoto N. Masuno,M. s Elk Grove, s 2015/02668432015,0203462 A1 9/20157/2015 CahanaBrowning et etal. al. CA (US); Ryan L. Smith, Sacramento, 2016/0002190 A1 1/2016 Browning et al. CA (US); John Bissell, II, Sacramento, 2016,01681 07 A1 6/2016 Masuno et al. CA (US) 2016/02O7897 A1 7, 2016 Wood et al. (73) Assignee: MICROMIDAS, INC., West FOREIGN PATENT DOCUMENTS Sacramento, CA (US) CN 101475544. A T 2009 (*) Notice: Subject to any disclaimer, the term of this N 1939: A 658 patent is extended or adjusted under 35 DE 635,783 C 9, 1936 U.S.C. 154(b) by 0 days. EP 291494 A2 11, 1988 EP 1049657 B1 3, 2003 (21) Appl. No.: 14/852,306 GB 1220851 A 1, 1971 GB 1448489 A 9, 1976 RU 2429234 C2 9, 2011 (22) Filed: Sep. -

Laboratory Rectifying Stills of Glass 2
» LABORATORY RECTIFYING STILLS OF GLASS 2 By Johannes H. Bruun 3 and Sylvester T. Schicktanz 3 ABSTRACT A complete description is given of a set of all-glass rectifying stills, suitable for distillation at pressures ranging from atmospheric down to about 50 mm. The stills are provided with efficient bubbling-cap columns containing 30 to 60 plates. Adiabatic conditions around the column are maintained by surrounding it with a jacket provided with a series of independent electrical heating units. Suitable means are provided for adjusting or maintaining the reflux ratio at the top of the column. For the purpose of conveniently obtaining an accurate value of the true boiling points of the distillates a continuous boiling-point apparatus is incor- porated in the receiving system. An efficient still of the packed-column type for distillations under pressures less than 50 mm is also described. Methods of operation and efficiency tests are given for the stills. CONTENTS Page I. Introduction 852 II. General features of the still assembly 852 III. Still pot 853 IV. Filling tube and heater for the still pot 853 V. Rectifying column 856 1. General features 856 2. Large-size column 856 3. Medium-size column 858 4. Small-size column 858 VI. Column jacket 861 VII. Reflux regulator 861 1. With variable reflux ratio 861 2. With constant reflux ratio 863 VIII. The continuous boiling-point apparatus 863 IX. Condensers and receiver 867 X. The mounting of the still 867 XI. Manufacture and transportation of the bubbling-cap still 870 XII. Operation of the bubbling-cap still 870 XIII. -

Chapter 18 Ethers and Epoxides; Thiols and Sulfides Ethers
Chapter 18 Ethers and Epoxides; Thiols and Sulfides Ethers • Ethers (R–O–R’): – Organic derivatives of water, having two organic groups bonded to the same oxygen atom © 2016 Cengage Learning 2 NAMES AND PROPERTIES OF ETHERS 3 Nomenclature: Common Names • Simple ethers are named by identifying two organic substituents and adding the word ether – Name the groups in alphabetical order – Symmetrical: Use dialkyl or just alkyl © 2016 Cengage Learning 4 Nomenclature: IUPAC Names • The more complex alkyl group is the parent name • The group with the oxygen becomes an alkoxy group © 2016 Cengage Learning 5 Nomenclature: Cyclic Ethers (Heterocycles) • Heterocyclic: Oxygen is part of the ring. O • Epoxides (oxiranes) H2C CH2 O • Oxetanes • Furans (Oxolanes) O O • Pyrans (Oxanes) O O O • Dioxanes O © 2013 Pearson Education, Inc. 6 Epoxide Nomenclature • Name the starting alkene and add “oxide” © 2013 Pearson Education, Inc. 7 Epoxide Nomenclature • The oxygen can be treated as a substituent (epoxy) on the compound • Use numbers to specify position • Oxygen is 1, the carbons are 2 and 3 • Substituents are named in alphabetical order © 2013 Pearson Education, Inc. 8 Properties of Ethers • Possess nearly the same geometry as water – Oxygen atom is sp3-hybridized – Bond angles of R–O–R bonds are approximately tetrahedral • Polar C—O bonds © 2013 Pearson Education, Inc. 9 Properties of Ethers: Hydrogen Bond • Hydrogen bond is a attractive interaction between an electronegative atom and a hydrogen atom bonded to another electronegative atom • Ethers cannot hydrogen bond with other ether molecules, so they have a lower boiling point than alcohols • Ether molecules can hydrogen bond with water and alcohol molecules • They are hydrogen bond acceptors © 2013 Pearson Education, Inc. -

Advices for Studying Organic Chemistry
Table of Contents Partial table of contents: Carbon Compounds and Chemical Bonds. Representative Carbon Compounds. An Introduction to Organic Reactions: Acids and Bases. Alkanes and Cycloalkanes: Conformations of Molecules. Stereochemistry: Chiral Molecules. Alkenes and Alkynes I: Properties and Synthesis. Alkenes and Alkynes II: Addition Reactions. Radical Reactions. Alcohols and Ethers. Conjugated Unsaturated Systems. Aromatic Compounds. Reactions of Aromatic Compounds. Aldehydes and Ketones I: Nucleophilic Additions to the Carbonyl Group. Aldehydes and Ketones II: Aldol Reactions. Carboxylic Acids and Their Derivatives: Nucleophilic Substitution at the Acyl Carbon. Amines. Carbohydrates. Lipids. Answers to Selected Problems. Glossary. Index. Solomons/Advices ADVICES FOR STUDYING ORGANIC CHEMISTRY 1. Keep up with your studying day to day –– never let yourself get behind, or better yet, be a little ahead of your instructor. Organic chemistry is a course in which one idea almost always builds on another that has gone before. 2. Study materials in small units, and be sure that you understand each new section before you go on to the next. Because of the cumulative nature of organic chemistry, your studying will be much more effective if you take each new idea as it comes and try to understand it completely before you move onto the nest concept. 3. Work all of the in-chapter and assigned problems. 4. Write when you study. Write the reactions, mechanisms, structures, and so on, over and over again. You need to know the material so thoroughly that you can explain it to someone else. This level of understanding comes to most of us (those of us without photographic memories) through writing. -
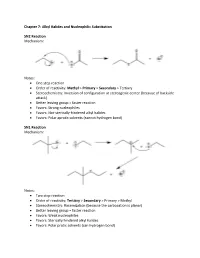
Alkyl Halides and Nucleophilic Substitution SN2 Reaction
Chapter 7: Alkyl Halides and Nucleophilic Substitution SN2 Reaction Mechanism: Notes: • One step reaction • Order of reactivity: Methyl > Primary > Secondary > Tertiary • Stereochemistry: Inversion of configuration at stereogenic center (because of backside attack) • Better leaving group = faster reaction • Favors: Strong nucleophiles • Favors: Not-sterically-hindered alkyl halides • Favors: Polar aprotic solvents (cannot hydrogen bond) SN1 Reaction Mechanism: Notes: • Two step reaction • Order of reactivity: Tertiary > Secondary > Primary > Methyl • Stereochemistry: Racemization (because the carbocation is planar) • Better leaving group = faster reaction • Favors: Weak nucleophiles • Favors: Sterically hindered alkyl halides • Favors: Polar protic solvents (can hydrogen bond) Important Trends Chapter 8: Alkyl Halides and Elimination Reactions E2 Reaction Mechanism: Notes: • One step reaction • Order of reactivity: Tertiary > Secondary > Primary • Stereochemistry: antiperiplanar arrangement of H and X • Better leaving group = faster reaction • Favors: Polar aprotic solvents, strong bases • Products follow Zaitsev rule (more substituted alkene is the major product) E1 Reaction Mechanism: Notes: • Two step reaction • Order of reactivity: Tertiary > Secondary > Primary • Stereochemistry: Trigonal planar carbocation intermediate • Better leaving group = faster reaction • Favors: Polar protic solvents, weak bases • Products follow Zaitsev rule Chapter 9: Alcohols, Ethers, and Epoxides Preparation of Alcohols Mechanism: Notes: • SN2 mechanism -

Reactions of Alkenes Since Bonds Are Stronger Than Bonds, Double Bonds Tend to React to Convert the Double Bond Into Bonds
Reactions of Alkenes Since bonds are stronger than bonds, double bonds tend to react to convert the double bond into bonds This is an addition reaction. (Other types of reaction have been substitution and elimination). Addition reactions are typically exothermic. Electrophilic Addition The bond is localized above and below the C-C bond. The electrons are relatively far away from the nuclei and are therefore loosely bound. An electrophile will attract those electrons, and can pull them away to form a new bond. This leaves one carbon with only 3 bonds and a +ve charge (carbocation). The double bond acts as a nucleophile (attacks the electrophile). Ch08 Reacns of Alkenes (landscape) Page 1 In most cases, the cation produced will react with another nucleophile to produce the final overall electrophilic addition product. Electrophilic addition is probably the most common reaction of alkenes. Consider the electrophilic addition of H-Br to but-2-ene: The alkene abstracts a proton from the HBr, and a carbocation and bromide ion are generated. The bromide ion quickly attacks the cationic center and yields the final product. In the final product, H-Br has been added across the double bond. Ch08 Reacns of Alkenes (landscape) Page 2 Orientation of Addition Consider the addition of H-Br to 2-methylbut-2-ene: There are two possible products arising from the two different ways of adding H-Br across the double bond. But only one is observed. The observed product is the one resulting from the more stable carbocation intermediate. Tertiary carbocations are more stable than secondary. -

The Claisen Condensation
Lecture 19 The Claisen Condensation •• •• • • O • O • – • CH3COCH2CH3 • CH2 COCH2CH3 March 29, 2016 Chemistry 328N Exam Tomorrow Evening!! Review tonight 5PM -6PM Welch 1.316 Chemistry 328N Some “loose ends” before we go on Spectrosopy of acid derivatives A selective reduction for your tool box Chemistry 328N Reduction of Acid Derivatives Acids (page 679-681) Esters (page 738-739) Please work through the example on 738 Amides (page 739-742) Nitriles (page 742) Selective reductions with NaBH4 Chemistry 328N DIBAlH Diisobutylaluminum hydride (DIBAlH) at -78°C selectively reduces esters to aldehydes •at -78°C, the tetrahedral intermediate does not collapse and it is not until hydrolysis in aqueous acid that the carbonyl group of the aldehyde is liberated Stable at low temperature Chemistry 328N Infrared Spectroscopy C=O stretching frequency depends on whether the compound is an acyl chloride, anhydride, ester, or amide. C=O stretching frequency n O O O O O CH3CCl CH3COCCH3 CH3COCH3 CH3CNH2 1822 cm-1 1748 1736 cm-1 1694 cm-1 and 1815 cm-1 Chemistry 328N Infrared Spectroscopy Anhydrides have two peaks due to C=O stretching. One from symmetrical stretching of the C=O and the other from an antisymmetrical stretch. C=O stretching frequency n O O CH3COCCH3 1748 and 1815 cm-1 Chemistry 328N Infrared Spectroscopy Nitriles are readily identified by absorption due to carbon-nitrogen triple bond stretching that is “all alone” in the 2210-2260 cm-1 region. Chemistry 328N Hydrolysis and Decarboxylation Chemistry 328N t-Butyl esters Chemistry 328N t-Butyl esters Chemistry 328N t-Butyl ester hydrolysis Note which bond is broken in this hydrolysis !! Chemistry 328N Recall our discussion of the acidity of protons a to carbonyls The anion is stabilized by resonance The better the stabilization, the more acidic the a proton Acidity of a protons on“normal” aldehydes and ketones is about that of alcohols and less than water…pKa ~ 18-20 Some are far more acidic, i.e.