Advices for Studying Organic Chemistry
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Organic Chemistry
Wisebridge Learning Systems Organic Chemistry Reaction Mechanisms Pocket-Book WLS www.wisebridgelearning.com © 2006 J S Wetzel LEARNING STRATEGIES CONTENTS ● The key to building intuition is to develop the habit ALKANES of asking how each particular mechanism reflects Thermal Cracking - Pyrolysis . 1 general principles. Look for the concepts behind Combustion . 1 the chemistry to make organic chemistry more co- Free Radical Halogenation. 2 herent and rewarding. ALKENES Electrophilic Addition of HX to Alkenes . 3 ● Acid Catalyzed Hydration of Alkenes . 4 Exothermic reactions tend to follow pathways Electrophilic Addition of Halogens to Alkenes . 5 where like charges can separate or where un- Halohydrin Formation . 6 like charges can come together. When reading Free Radical Addition of HX to Alkenes . 7 organic chemistry mechanisms, keep the elec- Catalytic Hydrogenation of Alkenes. 8 tronegativities of the elements and their valence Oxidation of Alkenes to Vicinal Diols. 9 electron configurations always in your mind. Try Oxidative Cleavage of Alkenes . 10 to nterpret electron movement in terms of energy Ozonolysis of Alkenes . 10 Allylic Halogenation . 11 to make the reactions easier to understand and Oxymercuration-Demercuration . 13 remember. Hydroboration of Alkenes . 14 ALKYNES ● For MCAT preparation, pay special attention to Electrophilic Addition of HX to Alkynes . 15 Hydration of Alkynes. 15 reactions where the product hinges on regio- Free Radical Addition of HX to Alkynes . 16 and stereo-selectivity and reactions involving Electrophilic Halogenation of Alkynes. 16 resonant intermediates, which are special favor- Hydroboration of Alkynes . 17 ites of the test-writers. Catalytic Hydrogenation of Alkynes. 17 Reduction of Alkynes with Alkali Metal/Ammonia . 18 Formation and Use of Acetylide Anion Nucleophiles . -

Reactions of Aromatic Compounds Just Like an Alkene, Benzene Has Clouds of Electrons Above and Below Its Sigma Bond Framework
Reactions of Aromatic Compounds Just like an alkene, benzene has clouds of electrons above and below its sigma bond framework. Although the electrons are in a stable aromatic system, they are still available for reaction with strong electrophiles. This generates a carbocation which is resonance stabilized (but not aromatic). This cation is called a sigma complex because the electrophile is joined to the benzene ring through a new sigma bond. The sigma complex (also called an arenium ion) is not aromatic since it contains an sp3 carbon (which disrupts the required loop of p orbitals). Ch17 Reactions of Aromatic Compounds (landscape).docx Page1 The loss of aromaticity required to form the sigma complex explains the highly endothermic nature of the first step. (That is why we require strong electrophiles for reaction). The sigma complex wishes to regain its aromaticity, and it may do so by either a reversal of the first step (i.e. regenerate the starting material) or by loss of the proton on the sp3 carbon (leading to a substitution product). When a reaction proceeds this way, it is electrophilic aromatic substitution. There are a wide variety of electrophiles that can be introduced into a benzene ring in this way, and so electrophilic aromatic substitution is a very important method for the synthesis of substituted aromatic compounds. Ch17 Reactions of Aromatic Compounds (landscape).docx Page2 Bromination of Benzene Bromination follows the same general mechanism for the electrophilic aromatic substitution (EAS). Bromine itself is not electrophilic enough to react with benzene. But the addition of a strong Lewis acid (electron pair acceptor), such as FeBr3, catalyses the reaction, and leads to the substitution product. -

Reactions of Alkenes and Alkynes
05 Reactions of Alkenes and Alkynes Polyethylene is the most widely used plastic, making up items such as packing foam, plastic bottles, and plastic utensils (top: © Jon Larson/iStockphoto; middle: GNL Media/Digital Vision/Getty Images, Inc.; bottom: © Lakhesis/iStockphoto). Inset: A model of ethylene. KEY QUESTIONS 5.1 What Are the Characteristic Reactions of Alkenes? 5.8 How Can Alkynes Be Reduced to Alkenes and 5.2 What Is a Reaction Mechanism? Alkanes? 5.3 What Are the Mechanisms of Electrophilic Additions HOW TO to Alkenes? 5.1 How to Draw Mechanisms 5.4 What Are Carbocation Rearrangements? 5.5 What Is Hydroboration–Oxidation of an Alkene? CHEMICAL CONNECTIONS 5.6 How Can an Alkene Be Reduced to an Alkane? 5A Catalytic Cracking and the Importance of Alkenes 5.7 How Can an Acetylide Anion Be Used to Create a New Carbon–Carbon Bond? IN THIS CHAPTER, we begin our systematic study of organic reactions and their mecha- nisms. Reaction mechanisms are step-by-step descriptions of how reactions proceed and are one of the most important unifying concepts in organic chemistry. We use the reactions of alkenes as the vehicle to introduce this concept. 129 130 CHAPTER 5 Reactions of Alkenes and Alkynes 5.1 What Are the Characteristic Reactions of Alkenes? The most characteristic reaction of alkenes is addition to the carbon–carbon double bond in such a way that the pi bond is broken and, in its place, sigma bonds are formed to two new atoms or groups of atoms. Several examples of reactions at the carbon–carbon double bond are shown in Table 5.1, along with the descriptive name(s) associated with each. -

Chapter 18 Ethers and Epoxides; Thiols and Sulfides Ethers
Chapter 18 Ethers and Epoxides; Thiols and Sulfides Ethers • Ethers (R–O–R’): – Organic derivatives of water, having two organic groups bonded to the same oxygen atom © 2016 Cengage Learning 2 NAMES AND PROPERTIES OF ETHERS 3 Nomenclature: Common Names • Simple ethers are named by identifying two organic substituents and adding the word ether – Name the groups in alphabetical order – Symmetrical: Use dialkyl or just alkyl © 2016 Cengage Learning 4 Nomenclature: IUPAC Names • The more complex alkyl group is the parent name • The group with the oxygen becomes an alkoxy group © 2016 Cengage Learning 5 Nomenclature: Cyclic Ethers (Heterocycles) • Heterocyclic: Oxygen is part of the ring. O • Epoxides (oxiranes) H2C CH2 O • Oxetanes • Furans (Oxolanes) O O • Pyrans (Oxanes) O O O • Dioxanes O © 2013 Pearson Education, Inc. 6 Epoxide Nomenclature • Name the starting alkene and add “oxide” © 2013 Pearson Education, Inc. 7 Epoxide Nomenclature • The oxygen can be treated as a substituent (epoxy) on the compound • Use numbers to specify position • Oxygen is 1, the carbons are 2 and 3 • Substituents are named in alphabetical order © 2013 Pearson Education, Inc. 8 Properties of Ethers • Possess nearly the same geometry as water – Oxygen atom is sp3-hybridized – Bond angles of R–O–R bonds are approximately tetrahedral • Polar C—O bonds © 2013 Pearson Education, Inc. 9 Properties of Ethers: Hydrogen Bond • Hydrogen bond is a attractive interaction between an electronegative atom and a hydrogen atom bonded to another electronegative atom • Ethers cannot hydrogen bond with other ether molecules, so they have a lower boiling point than alcohols • Ether molecules can hydrogen bond with water and alcohol molecules • They are hydrogen bond acceptors © 2013 Pearson Education, Inc. -

Aromatic Substitution Reactions: an Overview
Review Article Published: 03 Feb, 2020 SF Journal of Pharmaceutical and Analytical Chemistry Aromatic Substitution Reactions: An Overview Kapoor Y1 and Kumar K1,2* 1School of Pharmaceutical Sciences, Apeejay Stya University, Sohna-Palwal Road, Sohna, Gurgaon, Haryana, India 2School of Pharmacy and Technology Management, SVKM’s NMIMS University, Hyderabad, Telangana, India Abstract The introduction or replacement of substituent’s on aromatic rings by substitution reactions is one of the most fundamental transformations in organic chemistry. On the basis of the reaction mechanism, these substitution reactions can be divided into electrophilic, nucleophilic, radical, and transition metal catalyzed. This article also focuses on electrophilic and nucleophilic substitution mechanisms. Introduction The replacement of an atom, generally hydrogen, or a group attached to the carbon from the benzene ring by another group is known as aromatic substitution. The regioselectivity of these reactions depends upon the nature of the existing substituent and can be ortho, Meta or Para selective. Electrophilic Aromatic Substitution (EAS) reactions are important for synthetic purposes and are also among the most thoroughly studied classes of organic reactions from a mechanistic point of view. A wide variety of electrophiles can effect aromatic substitution. Usually, it is a substitution of some other group for hydrogen that is of interest, but this is not always the case. For example, both silicon and mercury substituent’s can be replaced by electrophiles. The reactivity of a particular electrophile determines which aromatic compounds can be successfully substituted. Despite the wide range of electrophilic species and aromatic ring systems that can undergo substitution, a single broad mechanistic picture encompasses most EAS reactions. -

Copyright © 2010 by Shauna M. Paradine ASYMMETRIC HALOFUNCTIONALIZATION of OLEFINS Reported by Shauna M. Paradine April 12
ASYMMETRIC HALOFUNCTIONALIZATION OF OLEFINS Reported by Shauna M. Paradine April 12, 2010 INTRODUCTION The functionalization of olefins is a commonly employed strategy for the rapid construction of molecular complexity in organic synthesis. Since the hybridization change from sp2 to sp3 at the reactive carbon atoms creates new stereocenters, performing these reactions in a well-controlled and highly asymmetric manner is ideal. Development of highly enantioselective olefin functionalization reactions has been a major focus of organic methodology for the past thirty years, and a number of useful and general methods have emerged for the asymmetric epoxidation, dioxygenation, aminooxygenation, hydrogenation, and hydroboration of olefins.1 Although halogen functionality is not as ubiquitous as oxygen and nitrogen in natural products, halogenated natural products still represent a pharmacologically important subclass2 and halogens themselves can be useful for further functionalization.3 The asymmetric addition of halogen functionality onto a carbon framework, therefore, has been of recent interest in synthetic methodology development. Synthetically useful asymmetric methods for the α–halogenation of carbonyls have been developed in response to this challenge,4 but general methods for the highly enantioselective halofunctionalization of olefins has thus far eluded organic chemists. Over the past few years, a number of research groups have developed methods seeking to fill this clear gap in asymmetric synthesis. This recent progress will be presented here. MECHANISTIC AND STEREOCHEMICAL CONSIDERATIONS The electrophilic halogenation of olefins is one of the oldest and most basic transformations in organic synthesis. In fact, “olefin”, which means “oil-forming”, was a term used to describe compounds, typically low molecular weight gaseous alkenes, that became oily liquids upon chlorination. -
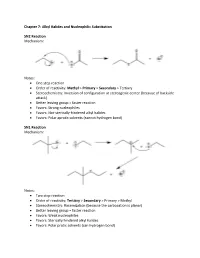
Alkyl Halides and Nucleophilic Substitution SN2 Reaction
Chapter 7: Alkyl Halides and Nucleophilic Substitution SN2 Reaction Mechanism: Notes: • One step reaction • Order of reactivity: Methyl > Primary > Secondary > Tertiary • Stereochemistry: Inversion of configuration at stereogenic center (because of backside attack) • Better leaving group = faster reaction • Favors: Strong nucleophiles • Favors: Not-sterically-hindered alkyl halides • Favors: Polar aprotic solvents (cannot hydrogen bond) SN1 Reaction Mechanism: Notes: • Two step reaction • Order of reactivity: Tertiary > Secondary > Primary > Methyl • Stereochemistry: Racemization (because the carbocation is planar) • Better leaving group = faster reaction • Favors: Weak nucleophiles • Favors: Sterically hindered alkyl halides • Favors: Polar protic solvents (can hydrogen bond) Important Trends Chapter 8: Alkyl Halides and Elimination Reactions E2 Reaction Mechanism: Notes: • One step reaction • Order of reactivity: Tertiary > Secondary > Primary • Stereochemistry: antiperiplanar arrangement of H and X • Better leaving group = faster reaction • Favors: Polar aprotic solvents, strong bases • Products follow Zaitsev rule (more substituted alkene is the major product) E1 Reaction Mechanism: Notes: • Two step reaction • Order of reactivity: Tertiary > Secondary > Primary • Stereochemistry: Trigonal planar carbocation intermediate • Better leaving group = faster reaction • Favors: Polar protic solvents, weak bases • Products follow Zaitsev rule Chapter 9: Alcohols, Ethers, and Epoxides Preparation of Alcohols Mechanism: Notes: • SN2 mechanism -

Halogen Bonds of Halonium Ions
Chem Soc Rev View Article Online REVIEW ARTICLE View Journal | View Issue Halogen bonds of halonium ions Cite this: Chem. Soc. Rev., 2020, Lotta Turunen and Ma´te´ Erde´lyi * 49,2688 Due to their electron deficiency, halonium ions act as particularly strong halogen bond donors. By accepting electrons in both lobes of their empty p-orbital, they are capable of simultaneously interacting with two Lewis bases. The interaction presumes the formation of three molecular orbitals and is accordingly typically entitled as a three-center halogen bond. In analogy to the [D–H–D]+ hydrogen bonds, which are at times entitled as short and strong bonds, the [D–X–D]+ halogen bonds of halonium ions show Bondi normalized interatomic distances of 0.6–0.7 and possess both charge transfer and electrostatic characteristics. The three-center halogen bond of halonium ions shows distinct differences in its properties from coordinative bonds of transition metals and is therefore Received 15th January 2020 applicable as a complementary synthon in supramolecular chemistry. The three-center halogen bond DOI: 10.1039/d0cs00034e modulates the reactivity of halonium ions and is hence a useful tool for synthetic organic chemistry. Following the discussion of the nature and properties of halonium ions’ halogen bonds, this tutorial Creative Commons Attribution 3.0 Unported Licence. rsc.li/chem-soc-rev review provides an overview of their current applications to stimulate future developments. Introduction resembles hydrogen bonding and has accordingly been proposed to be applicable as a complementary tool for Halogen bonding (XB) is the directional, non-covalent interaction the rational modulation of molecular recognition events in of an electron-poor region of a halogen and a Lewis base.1 It chemistry and biology.2 Being a weak interaction, halogen bonding is typically difficult to detect, especially in disordered This article is licensed under a 3 Department of Chemistry – BMC, Uppsala University, SE-751 23 Uppsala, Sweden. -

Reactions of Alkenes Since Bonds Are Stronger Than Bonds, Double Bonds Tend to React to Convert the Double Bond Into Bonds
Reactions of Alkenes Since bonds are stronger than bonds, double bonds tend to react to convert the double bond into bonds This is an addition reaction. (Other types of reaction have been substitution and elimination). Addition reactions are typically exothermic. Electrophilic Addition The bond is localized above and below the C-C bond. The electrons are relatively far away from the nuclei and are therefore loosely bound. An electrophile will attract those electrons, and can pull them away to form a new bond. This leaves one carbon with only 3 bonds and a +ve charge (carbocation). The double bond acts as a nucleophile (attacks the electrophile). Ch08 Reacns of Alkenes (landscape) Page 1 In most cases, the cation produced will react with another nucleophile to produce the final overall electrophilic addition product. Electrophilic addition is probably the most common reaction of alkenes. Consider the electrophilic addition of H-Br to but-2-ene: The alkene abstracts a proton from the HBr, and a carbocation and bromide ion are generated. The bromide ion quickly attacks the cationic center and yields the final product. In the final product, H-Br has been added across the double bond. Ch08 Reacns of Alkenes (landscape) Page 2 Orientation of Addition Consider the addition of H-Br to 2-methylbut-2-ene: There are two possible products arising from the two different ways of adding H-Br across the double bond. But only one is observed. The observed product is the one resulting from the more stable carbocation intermediate. Tertiary carbocations are more stable than secondary. -

Halogen Bonds in Solution Cite This: DOI: 10.1039/X0xx00000x Alavi Karim, Marcus Reitti, Anna�Carin C
Chemical Science Accepted Manuscript This is an Accepted Manuscript, which has been through the Royal Society of Chemistry peer review process and has been accepted for publication. Accepted Manuscripts are published online shortly after acceptance, before technical editing, formatting and proof reading. Using this free service, authors can make their results available to the community, in citable form, before we publish the edited article. We will replace this Accepted Manuscript with the edited and formatted Advance Article as soon as it is available. You can find more information about Accepted Manuscripts in the Information for Authors. Please note that technical editing may introduce minor changes to the text and/or graphics, which may alter content. The journal’s standard Terms & Conditions and the Ethical guidelines still apply. In no event shall the Royal Society of Chemistry be held responsible for any errors or omissions in this Accepted Manuscript or any consequences arising from the use of any information it contains. www.rsc.org/chemicalscience Page 1 of 8 Chemical Science Chemical Science RSC Publishing ARTICLE The nature of [N −−−Cl −−−N] + and [N −−−F−−−N] + halogen bonds in solution Cite this: DOI: 10.1039/x0xx00000x Alavi Karim, Marcus Reitti, Anna-Carin C. Carlsson, Jürgen Gräfenstein and Máté Erdélyi * Received 00th January 2012, Accepted 00th January 2012 Halonium ions are synthetically useful, transient species that may be stabilized by attachment to two electron donors. Whereas studies of [C −X−C] +-type ions have greatly contributed to the DOI: 10.1039/x0xx00000x fundamental understanding of chemical bonding and reaction mechanisms, investigations of + www.rsc.org/ the corresponding [N −X−N] halogen bond complexes are only at an early stage. -

Reaction-Of-Alkenes.Pdf
8 Reactions of Alkenes Goals for Chapter 8 1 Explain why electrophilic additions are among the most common reactions of alkenes. femoral component 2 Predict the products of the reactions of alkenes, including the orientation of the reaction (regio- polyethylene bearing chemistry) and the stereochemistry. tibial plate 3 Propose mechanisms to explain the observed products of alkene reactions. 4 Use retrosynthetic analysis to solve multistep synthesis problems with alkenes as reagents, intermediates, or products. ◀ Polyethylene provides a self-lubricating surface for movement of the metal parts of an artificial knee replacement. The highly cross-linked polyethylene used in these implants is fabricated to be exceptionally tough: It wears only about 0.1 mm per year. Polyethylene is compatible with the human body, and in most cases, it does not cause a foreign body reaction even after years of constant movement in the joint. The alkene double bond is a gateway functional group. Alkene reactions lead to many other functional groups that lay the foundation for the rest of your study of organic chemistry. You can convert alkenes to alkyl halides, epoxides, alcohols, aldehydes, ketones, carboxylic acids, and other functional groups. The reactions of alkenes arise from the reactivity of their carbon–carbon double bonds. Organic chemists enjoy the challenge of taking a simple carbon–carbon double bond and manipulating it in all possible ways to produce other compounds, often mimicking biological reactions that occur in cells. This chapter covers the most common alkene reactions, including their mechanisms, reactivity, orientation, and stereochemistry. Most reactions of alkenes involve addition of atoms or groups across the double bond, with one atom or group adding to each end. -

Reactions of Alkenes
Reactions of Alkenes Alkenes generally react in an addition mechanism (addition – two new species add to a molecule and none leave) R X Y X Y H R R R H Have already observed using a H+ electrophile (HBr or H+/H2O) that a carbocation intermediate is generated which directs the regiochemistry Whenever a free carbocation intermediate is generated there will not be a stereopreference due to the nucleophile being able to react on either lobe of the carbocation (already observed this with SN1 and E1 reactions) Br Br H+ H H3C Br CH2CH3 Obtain racemic mixture of this regioisomer Reactions of Alkenes There are three questions to ask for any addition reaction R X Y X Y H R R R H 1) What is being added? (what is the electrophile?) 2) What is the regiochemistry? (do the reagents add with the X group to the left or right?) 3) What is the stereochemistry? (do both the X and Y groups add to the same side of the double bond or opposite sides?) All of these questions can be answered if the intermediate structure is known Reactions of Alkenes Dihalogen compounds can also react as electrophiles in reactions with alkenes Possible partial bond structures + !+ Br Br ! Br Br Br !+ or Br !+ More stable partial positive charge Experimentally it is known, however, that rearrangements do nor occur with Br2 addition -therefore free carbocations must not be present The large size and polarizability of the halogen can stabilize the unstable carbocation With an unsymmetrical alkene, however, both bonds to the bromine need not be equivalent Called a “Bromonium” ion