PDF Hosted at the Radboud Repository of the Radboud University Nijmegen
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

TGF-Β1 Signaling Targets Metastasis-Associated Protein 1, a New Effector in Epithelial Cells
Oncogene (2011) 30, 2230–2241 & 2011 Macmillan Publishers Limited All rights reserved 0950-9232/11 www.nature.com/onc ORIGINAL ARTICLE TGF-b1 signaling targets metastasis-associated protein 1, a new effector in epithelial cells SB Pakala1, K Singh1,3, SDN Reddy1, K Ohshiro1, D-Q Li1, L Mishra2 and R Kumar1 1Department of Biochemistry and Molecular Biology and Institute of Coregulator Biology, The George Washington University Medical Center, Washington, DC, USA and 2Department of Gastroenterology, Hepatology and Nutrition, The University of Texas MD Anderson Cancer Center, Houston, TX, USA In spite of a large number of transforming growth factor b1 gene chromatin in response to upstream signals. The (TGF-b1)-regulated genes, the nature of its targets with TGF-b1-signaling is largely mediated by Smad proteins roles in transformation continues to be poorly understood. (Massague et al., 2005) where Smad2 and Smad3 are Here, we discovered that TGF-b1 stimulates transcription phosphorylated by TGF-b1-receptors and associate with of metastasis-associated protein 1 (MTA1), a dual master the common mediator Smad4, which translocates to the coregulator, in epithelial cells, and that MTA1 status is a nucleus to participate in the expression of TGF-b1-target determinant of TGF-b1-induced epithelial-to-mesenchymal genes (Deckers et al., 2006). Previous studies have shown transition (EMT) phenotypes. In addition, we found that that CUTL1, also known as CDP (CCAAT displacement MTA1/polymerase II/activator protein-1 (AP-1) co-activator protein), a target of TGF-b1, is needed for its short-term complex interacts with the FosB-gene chromatin and stimu- effects of TGF-b1 on cell motility involving Smad4- lates its transcription, and FosB in turn, utilizes FosB/histone dependent pathway (Michl et al.,2005). -

Micrornas in Cardiovascular Disease: from Pathogenesis to Prevention and Treatment
MicroRNAs in cardiovascular disease: from pathogenesis to prevention and treatment Daniel Quiat, Eric N. Olson J Clin Invest. 2013;123(1):11-18. https://doi.org/10.1172/JCI62876. Review Series The management of cardiovascular risk through lifestyle modification and pharmacotherapy is paramount to the prevention of cardiovascular disease. Epidemiological studies have identified obesity, dyslipidemia, diabetes, and hypertension as interrelated factors that negatively affect cardiovascular health. Recently, genetic and pharmacological evidence in model systems has implicated microRNAs as dynamic modifiers of disease pathogenesis. An expanded understanding of the function of microRNAs in gene regulatory networks associated with cardiovascular risk will enable identification of novel genetic mechanisms of disease and inform the development of innovative therapeutic strategies. Find the latest version: https://jci.me/62876/pdf Review series MicroRNAs in cardiovascular disease: from pathogenesis to prevention and treatment Daniel Quiat and Eric N. Olson Department of Molecular Biology, University of Texas Southwestern Medical Center, Dallas, Texas, USA. The management of cardiovascular risk through lifestyle modification and pharmacotherapy is paramount to the prevention of cardiovascular disease. Epidemiological studies have identified obesity, dyslipidemia, diabetes, and hypertension as interrelated factors that negatively affect cardiovascular health. Recently, genetic and pharmaco- logical evidence in model systems has implicated microRNAs -

I3C & Hormone Positive Breast Cancer
ARTICLE IN PRESS Journal of Nutritional Biochemistry xx (2006) xxx–xxx Estrogen receptor a as a target for indole-3-carbinol Thomas T.Y. Wanga,4, Matthew J. Milnerb, John A. Milnerc, Young S. Kimc aPhytonutrients Laboratory, Beltsville Human Nutrition Research Center, Agricultural Research Service, U.S. Department of Agriculture, Beltsville, MD 20705, USA bDepartment of Biology, The Pennsylvania State University, University Park, PA, USA cNutritional Science Research Group, Division of Cancer Prevention, National Cancer Institute, National Institutes of Health, Bethesda, MD, USA Received 8 June 2005; received in revised form 14 October 2005; accepted 22 October 2005 Abstract A wealth of preclinical evidence supports the antitumorigenic properties of indole-3-carbinol (I3C), which is a major bioactive food component in cruciferous vegetables. However, the underlying molecular mechanism(s) accounting for these effects remain unresolved. In the present study, estrogen receptor alpha (ER-a) was identified as a potential molecular target for I3C. Treating MCF-7 cells with 100 AM I3C reduced ER-a mRNA expression by approximately 60% compared to controls. This reduction in ER-a transcript levels was confirmed using real-time polymerase chain reaction. The I3C dimer, 3,3V-diindolylmethane (DIM), was considerably more effective in depressing ER-a mRNA in MCF-7 cells than the monomeric unit. The suppressive effects of 5 AM DIM on ER-a mRNA was comparable to that caused by 100 AM I3C. DIM is known to accumulate in the nucleus and is a preferred ligand for aryl hydrocarbon receptor (AhR) to I3C. The addition of other AhR ligands, a-naphthoflavone (a-NF, 10 AM) and luteolin (10 AM), to the culture media resulted in a similar suppression in ER-a mRNA levels to that caused by 5 AM DIM. -
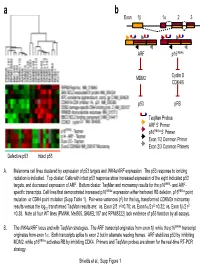
Taqman Probes ARF 5' Primer P16ink4a 5' Primer Exon 1/2
a b Exon 1β 1α 23 ARF p16INK4a MDM2 Cyclin D CDK4/6 p53 pRB TaqMan Probes ARF 5’ Primer p16INK4a 5’ Primer Exon 1/2 Common Primer Exon 2/3 Common Primers Defective p53 Intact p53 A. Melanoma cell lines clustered by expression of p53 targets and INK4a/ARF expression. The p53 response to ionizing radiation is indicated. Top cluster: Cells with intact p53 response show increased expression of the eight indicated p53 targets, and decreased expression of ARF. Bottom cluster: TaqMan and microarray results for the p16INK4- and ARF- specific transcripts. Cell lines that demonstrated increased p16INK4a expression either harbored RB deletion, p16INK4a point 2 mutation or CDK4 point mutation (Supp Table 1). Pair-wise variances (r ) for the log2 transformed CDKN2a microarray 2 2 2 results versus the log10 transformed TaqMan results are: vs. Exon 2/3 r =0.78; vs. Exon1α/2 r =0.32; vs. Exon 1β/2 r =0.38. Note all four WT lines (PMWK, Mel505, SKMEL187 and RPMI8322) lack evidence of p53 function by all assays. B. The INK4a/ARF locus and with TaqMan strategies. The ARF transcript originates from exon 1β while the p16INK4a transcript originates from exon 1α. Both transcripts splice to exon 2 but in alternate reading frames. ARF stabilizes p53 by inhibiting MDM2, while p16INK4a activates RB by inhibiting CDK4. Primers and TaqMan probes are shown for the real-time RT-PCR strategy. Shields et al., Supp Figure 1 SKMEL 28 U01 24h SKMEL WM2664 U01 48h WM2664 U01 24h 24 U01 48h SKMEL 24 U01 24h SKMEL 24 Untreated SKMEL 24 DMSO 48h SKMEL 24 DMSO 24h SKMEL WM2664 DMSO 24h WM2664 DMSO 48h WM2644 Untreated 28 Untreated SKMEL 28 DMSO 48h SKMEL 28 DMSO 24h SKMEL -3.00 -2.00 -1.00 0.00 1.00 2.00 3.00 relative to median expression Genes decreased by UO1 (863) HIF1A Hypoxia-inducible factor 1, alpha subunit NM_001530 RBBP8 Retinoblastoma binding protein 8 NM_002894 Homo sapiens, clone IMAGE:4337652, mRNA BC018676 EIF4EBP1 Eukaryotic translation initiation factor 4E binding protein EXOSC8 Exosome component 8 NM_181503 ENST00000321524 MCM7 MCM7 minichromosome maintenance deficient 7 (S. -

Transcriptional Mechanisms of WNT5A Based on NF-Κb, Hedgehog, Tgfß, and Notch Signaling Cascades
763-769 23/4/2009 01:21 ÌÌ Page 763 INTERNATIONAL JOURNAL OF MOLECULAR MEDICINE 23: 763-769, 2009 763 Transcriptional mechanisms of WNT5A based on NF-κB, Hedgehog, TGFß, and Notch signaling cascades MASUKO KATOH1 and MASARU KATOH2 1M&M Medical BioInformatics, Hongo 113-0033; 2Genetics and Cell Biology Section, National Cancer Center, Tokyo 104-0045, Japan Received March 5, 2009; Accepted April 2, 2009 DOI: 10.3892/ijmm_00000190 Abstract. WNT5A is a cancer-associated gene involved in were found to upregulate WNT5A expression directly through invasion and metastasis of melanoma, breast cancer, pancreatic the Smad complex, and also indirectly through Smad- cancer, and gastric cancer. WNT5A transduces signals through induced CUX1 and MAP3K7-mediated NF-κB. Together Frizzled, ROR1, ROR2 or RYK receptors to ß-catenin-TCF/ these facts indicate that WNT5A is transcribed based on LEF, DVL-RhoA-ROCK, DVL-RhoB-Rab4, DVL-Rac-JNK, multiple mechanisms, such as NF-κB, Hedgehog, TGFß, and DVL-aPKC, Calcineurin-NFAT, MAP3K7-NLK, MAP3K7- Notch signaling cascades. NF-κB, and DAG-PKC signaling cascades in a context- dependent manner. SNAI1 (Snail), CD44, G3BP2, and YAP1 Introduction are WNT5A target genes. We and other groups previously reported that IL6- or LIF-induced signaling through JAK- WNT signaling cascades are involved in a variety of cellular STAT3 signaling cascade is involved in WNT5A upregulation processes during embryogenesis and carcinogenesis (1-4). (STAT3-WNT5A signaling loop). Here, refined integrative Because biological functions of human genes and those of genomic analyses of WNT5A were carried out to elucidate model-animal orthologs are not always conserved due to other mechanisms of WNT5A transcription. -

Tumor Microenvironment: Driving Forces and Potential Therapeutic Targets for Breast Cancer Metastasis
Xie et al. Chin J Cancer (2017) 36:36 DOI 10.1186/s40880-017-0202-y Chinese Journal of Cancer REVIEW Open Access Tumor microenvironment: driving forces and potential therapeutic targets for breast cancer metastasis Hong‑Yan Xie1,2,3, Zhi‑Min Shao1,2,3,4,5* and Da‑Qiang Li1,2,3,4* Abstract Distant metastasis to specific target organs is responsible for over 90% of breast cancer-related deaths, but the underlying molecular mechanism is unclear. Mounting evidence suggests that the interplay between breast cancer cells and the target organ microenvironment is the key determinant of organ-specific metastasis of this lethal disease. Here, we highlight new findings and concepts concerning the emerging role of the tumor microenvironment in breast cancer metastasis; we also discuss potential therapeutic intervention strategies aimed at targeting compo‑ nents of the tumor microenvironment. Keywords: Breast cancer, Invasion and metastasis, Tumor microenvironment Background investigated extensively. Recently, several laboratories In terms of its molecular characteristics and clinical phe- screened and identified a batch of breast cancer metas- notypes, breast cancer is a highly heterogeneous malig- tasis-related genes using well-established animal mod- nancy. Like other solid tumor cells, breast cancer cells els of metastatic breast cancer in combination with tend to metastasize to distant organs, such as the lung [1, high-throughput genomic analysis (Table 1). For exam- 2], liver [2], bone [3], and brain [4], through local invasion ple, it has been -

Williams Syndrome As a Model for Elucidation of the Pathway Genes – the Brain – Cognitive Functions: Genetics and Epigenetics
reVIeWS Williams Syndrome As a Model for Elucidation of the Pathway Genes – the Brain – Cognitive Functions: Genetics and Epigenetics Е. А. Nikitina1,2*, A. V. Medvedeva1,3, G. А. Zakharov1,3, Е. V. Savvateeva-Popova1,3 1Pavlov Institute of Physiology, Russian Academy of Sciences, nab. Makarova, 6, 199034, St. Petersburg, Russia 2Herzen State Pedagogical University, nab. r. Moyki, 48, 191186, St. Petersburg, Russia 3Saint Petersburg State University, Universitetskaya nab., 8-9, 199034, St. Petersburg, Russia *E-mail: [email protected] Received 16.07.2013 Copyright © 2014 Park-media, Ltd. This is an open access article distributed under the Creative Commons Attribution License,which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. abStract Genomic diseases or syndromes with multiple manifestations arise spontaneously and unpredict- ably as a result of contiguous deletions and duplications generated by unequal recombination in chromosomal regions with a specific architecture. The Williams syndrome is believed to be one of the most attractive models for linking genes, the brain, behavior and cognitive functions. It is a neurogenetic disorder resulting from a 1.5 Mb deletion at 7q11.23 which covers more than 20 genes; the hemizigosity of these genes leads to multiple mani- festations, with the behavioral ones comprising three distinct domains: 1) visuo-spatial orientation; 2) verbal and linguistic defect; and 3) hypersocialisation. The shortest observed deletion leads to hemizigosity in only two genes: eln and limk1. Therefore, the first gene is supposed to be responsible for cardiovascular pathology; and the second one, for cognitive pathology. Since cognitive pathology diminishes with a patient’s age, the original idea of the crucial role of genes straightforwardly determining the brain’s morphology and behavior was substituted by ideas of the brain’s plasticity and the necessity of finding epigenetic factors that affect brain development and the functions manifested as behavioral changes. -

1 Novel Expression Signatures Identified by Transcriptional Analysis
ARD Online First, published on October 7, 2009 as 10.1136/ard.2009.108043 Ann Rheum Dis: first published as 10.1136/ard.2009.108043 on 7 October 2009. Downloaded from Novel expression signatures identified by transcriptional analysis of separated leukocyte subsets in SLE and vasculitis 1Paul A Lyons, 1Eoin F McKinney, 1Tim F Rayner, 1Alexander Hatton, 1Hayley B Woffendin, 1Maria Koukoulaki, 2Thomas C Freeman, 1David RW Jayne, 1Afzal N Chaudhry, and 1Kenneth GC Smith. 1Cambridge Institute for Medical Research and Department of Medicine, Addenbrooke’s Hospital, Hills Road, Cambridge, CB2 0XY, UK 2Roslin Institute, University of Edinburgh, Roslin, Midlothian, EH25 9PS, UK Correspondence should be addressed to Dr Paul Lyons or Prof Kenneth Smith, Department of Medicine, Cambridge Institute for Medical Research, Addenbrooke’s Hospital, Hills Road, Cambridge, CB2 0XY, UK. Telephone: +44 1223 762642, Fax: +44 1223 762640, E-mail: [email protected] or [email protected] Key words: Gene expression, autoimmune disease, SLE, vasculitis Word count: 2,906 The Corresponding Author has the right to grant on behalf of all authors and does grant on behalf of all authors, an exclusive licence (or non-exclusive for government employees) on a worldwide basis to the BMJ Publishing Group Ltd and its Licensees to permit this article (if accepted) to be published in Annals of the Rheumatic Diseases and any other BMJPGL products to exploit all subsidiary rights, as set out in their licence (http://ard.bmj.com/ifora/licence.pdf). http://ard.bmj.com/ on September 29, 2021 by guest. Protected copyright. 1 Copyright Article author (or their employer) 2009. -

Genetic and Functional Differences Between Multipotent Neural and Pluripotent Embryonic Stem Cells
Colloquium Genetic and functional differences between multipotent neural and pluripotent embryonic stem cells Kevin A. D’Amour and Fred H. Gage* Laboratory of Genetics, The Salk Institute for Biological Studies, 10010 North Torrey Pines Road, La Jolla, CA 92037 Stem cells (SCs) are functionally defined by their abilities to KSϩ clone of this region was the kind gift of Angie Rizzino (18). self-renew and generate differentiated cells. Although much effort Details regarding construction of the P͞Sox2-GFP-Ires-Puro has been focused on defining the common characteristics among vector are available on request. various types of SCs, the genetic and functional differences be- Transgenic ESC clones were derived from ROSA26-4 (19) tween multipotent and pluripotent SCs have garnered less atten- ESCs as described (20) and were microinjected into blastocysts tion. We report a direct genetic and functional comparison of to verify ESC clones that exhibited telencephalic-restricted molecularly defined and clonally related populations of neural SCs enhanced GFP (EGFP) expression. (NSCs) and embryonic SCs (ESCs), using the Sox2 promoter for Chimeric fetuses were generated via blastocyst injection of isolation of purified populations by fluorescence-activated cell P͞Sox2-GIP ESCs, collected at embryonic day (E) 14, and exam- sorting. A stringent expression profile comparison of promoter- ined by epifluorescent microscopy to select fetuses exhibiting high defined NSCs and ESCs revealed a striking dissimilarity, and sub- degrees of chimerism (Ն75%). The telencephalon was dissected sequent chimera analyses confirmed the fundamental differences free of the meninges and remaining brain tissue, diced with a scalpel in cellular potency between these populations. This direct com- blade, and digested for 5 min at 37°C in 1 ml of 0.25% wt/vol parison elucidates the molecular basis for the functional differ- trypsin͞0.54 mM EDTA. -

WO 2012/054896 Al
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number ι (43) International Publication Date ¾ ί t 2 6 April 2012 (26.04.2012) WO 2012/054896 Al (51) International Patent Classification: AO, AT, AU, AZ, BA, BB, BG, BH, BR, BW, BY, BZ, C12N 5/00 (2006.01) C12N 15/00 (2006.01) CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, C12N 5/02 (2006.01) DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KM, KN, KP, (21) International Application Number: KR, KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, PCT/US201 1/057387 ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, (22) International Filing Date: NO, NZ, OM, PE, PG, PH, PL, PT, QA, RO, RS, RU, 2 1 October 201 1 (21 .10.201 1) RW, SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, (25) Filing Language: English ZM, ZW. (26) Publication Language: English (84) Designated States (unless otherwise indicated, for every (30) Priority Data: kind of regional protection available): ARIPO (BW, GH, 61/406,064 22 October 2010 (22.10.2010) US GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, SZ, TZ, 61/415,244 18 November 2010 (18.1 1.2010) US UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, MD, RU, TJ, TM), European (AL, AT, BE, BG, CH, CY, CZ, (71) Applicant (for all designated States except US): BIO- DE, DK, EE, ES, FI, FR, GB, GR, HR, HU, IE, IS, IT, TIME INC. -

BMC Biology Biomed Central
BMC Biology BioMed Central Research article Open Access Classification and nomenclature of all human homeobox genes PeterWHHolland*†1, H Anne F Booth†1 and Elspeth A Bruford2 Address: 1Department of Zoology, University of Oxford, South Parks Road, Oxford, OX1 3PS, UK and 2HUGO Gene Nomenclature Committee, European Bioinformatics Institute (EMBL-EBI), Wellcome Trust Genome Campus, Hinxton, Cambridgeshire, CB10 1SA, UK Email: Peter WH Holland* - [email protected]; H Anne F Booth - [email protected]; Elspeth A Bruford - [email protected] * Corresponding author †Equal contributors Published: 26 October 2007 Received: 30 March 2007 Accepted: 26 October 2007 BMC Biology 2007, 5:47 doi:10.1186/1741-7007-5-47 This article is available from: http://www.biomedcentral.com/1741-7007/5/47 © 2007 Holland et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: The homeobox genes are a large and diverse group of genes, many of which play important roles in the embryonic development of animals. Increasingly, homeobox genes are being compared between genomes in an attempt to understand the evolution of animal development. Despite their importance, the full diversity of human homeobox genes has not previously been described. Results: We have identified all homeobox genes and pseudogenes in the euchromatic regions of the human genome, finding many unannotated, incorrectly annotated, unnamed, misnamed or misclassified genes and pseudogenes. -

Factor Expression and Correlate with Specific Transcription in Early Human Precursor B Cell Subsets Ig Gene Rearrangement Steps
The Journal of Immunology Ig Gene Rearrangement Steps Are Initiated in Early Human Precursor B Cell Subsets and Correlate with Specific Transcription Factor Expression1 Menno C. van Zelm,*† Mirjam van der Burg,* Dick de Ridder,*‡ Barbara H. Barendregt,*† Edwin F. E. de Haas,* Marcel J. T. Reinders,‡ Arjan C. Lankester,§ Tom Re´ve´sz,¶ Frank J. T. Staal,* and Jacques J. M. van Dongen2* The role of specific transcription factors in the initiation and regulation of Ig gene rearrangements has been studied extensively in mouse models, but data on normal human precursor B cell differentiation are limited. We purified five human precursor B cell subsets, and assessed and quantified their IGH, IGK, and IGL gene rearrangement patterns and gene expression profiles. Pro-B cells already massively initiate DH-JH rearrangements, which are completed with VH-DJH rearrangements in pre-B-I cells. Large cycling pre-B-II cells are selected for in-frame IGH gene rearrangements. The first IGK/IGL gene rearrangements were initiated in pre-B-I cells, but their frequency increased enormously in small pre-B-II cells, and in-frame selection was found in immature B cells. Transcripts of the RAG1 and RAG2 genes and earlier defined transcription factors, such as E2A, early B cell factor, E2-2, PAX5, and IRF4, were specifically up-regulated at stages undergoing Ig gene rearrangements. Based on the combined Ig gene rearrangement status and gene expression profiles of consecutive precursor B cell subsets, we identified 16 candidate genes involved in initiation and/or regulation of Ig gene rearrangements. These analyses provide new insights into early human pre- cursor B cell differentiation steps and represent an excellent template for studies on oncogenic transformation in precursor B acute lymphoblastic leukemia and B cell differentiation blocks in primary Ab deficiencies.