Williams Syndrome As a Model for Elucidation of the Pathway Genes – the Brain – Cognitive Functions: Genetics and Epigenetics
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Role of LIM Kinase 1 and Its Substrates in Cell Cycle Progression
University of Central Florida STARS Electronic Theses and Dissertations, 2004-2019 2014 The Role of LIM Kinase 1 and its Substrates in Cell Cycle Progression Lisa Ritchey University of Central Florida Part of the Medical Sciences Commons Find similar works at: https://stars.library.ucf.edu/etd University of Central Florida Libraries http://library.ucf.edu This Doctoral Dissertation (Open Access) is brought to you for free and open access by STARS. It has been accepted for inclusion in Electronic Theses and Dissertations, 2004-2019 by an authorized administrator of STARS. For more information, please contact [email protected]. STARS Citation Ritchey, Lisa, "The Role of LIM Kinase 1 and its Substrates in Cell Cycle Progression" (2014). Electronic Theses and Dissertations, 2004-2019. 1300. https://stars.library.ucf.edu/etd/1300 THE ROLE OF LIM KINASE 1 AND ITS SUBSTRATES IN CELL CYCLE PROGRESSION by LISA RITCHEY B.S. Florida State University 2007 M.S. University of Central Florida 2010 A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Burnett School of Biomedical Sciences in the College of Graduate Studies at the University of Central Florida Orlando, Florida Summer Term 2014 Major Professor: Ratna Chakrabarti © 2014 Lisa Ritchey ii ABSTRACT LIM Kinase 1 (LIMK1), a modulator of actin and microtubule dynamics, has been shown to be involved in cell cycle progression. In this study we examine the role of LIMK1 in G1 phase and mitosis. We found ectopic expression of LIMK1 resulted in altered expression of p27Kip1, the G1 phase Cyclin D1/Cdk4 inhibitor. -

The Role of P21-Activated Protein Kinase 1 in Metabolic Homeostasis
THE ROLE OF P21-ACTIVATED PROTEIN KINASE 1 IN METABOLIC HOMEOSTASIS by YU-TING CHIANG A thesis submitted in conformity with the requirements for the degree of Doctor of Philosophy Graduate Department of Physiology University of Toronto © Copyright by Yu-ting Chiang 2014 The Role of P21-Activated Protein Kinase 1 in Metabolic Homeostasis Yu-ting Chiang Doctor of Philosophy Department of Physiology University of Toronto 2014 Abstract Our laboratory has demonstrated previously that the proglucagon gene (gcg), which encodes the incretin hormone GLP-1, is among the downstream targets of the Wnt signaling pathway; and that Pak1 mediates the stimulatory effect of insulin on Wnt target gene expression in mouse gut non- endocrine cells. Here, I asked whether Pak1 controls gut gcg expression and GLP-1 production, and whether Pak1 deletion leads to impaired metabolic homeostasis in mice. I detected the expression of Pak1 and two other group I Paks in the gut endocrine L cell line GLUTag, and co- localized Pak1 and GLP-1 in the mouse gut. Insulin was shown to stimulate Pak1 Thr423 and β-cat Ser675 phosphorylation. The stimulation of insulin on β-cat Ser675 phosphorylation, gcg promoter activity and gcg mRNA expression could be attenuated by the Pak inhibitor IPA3. Male Pak1-/- mice showed significant reduction in both gut and brain gcg expression levels, and attenuated elevation of plasma GLP-1 levels in response to oral glucose challenge. Notably, the Pak1-/- mice were intolerant to both intraperitoneal and oral glucose administration. Aged Pak1-/- mice showed a severe defect in response to intraperitoneal pyruvate challenge (IPPTT). -

TGF-Β1 Signaling Targets Metastasis-Associated Protein 1, a New Effector in Epithelial Cells
Oncogene (2011) 30, 2230–2241 & 2011 Macmillan Publishers Limited All rights reserved 0950-9232/11 www.nature.com/onc ORIGINAL ARTICLE TGF-b1 signaling targets metastasis-associated protein 1, a new effector in epithelial cells SB Pakala1, K Singh1,3, SDN Reddy1, K Ohshiro1, D-Q Li1, L Mishra2 and R Kumar1 1Department of Biochemistry and Molecular Biology and Institute of Coregulator Biology, The George Washington University Medical Center, Washington, DC, USA and 2Department of Gastroenterology, Hepatology and Nutrition, The University of Texas MD Anderson Cancer Center, Houston, TX, USA In spite of a large number of transforming growth factor b1 gene chromatin in response to upstream signals. The (TGF-b1)-regulated genes, the nature of its targets with TGF-b1-signaling is largely mediated by Smad proteins roles in transformation continues to be poorly understood. (Massague et al., 2005) where Smad2 and Smad3 are Here, we discovered that TGF-b1 stimulates transcription phosphorylated by TGF-b1-receptors and associate with of metastasis-associated protein 1 (MTA1), a dual master the common mediator Smad4, which translocates to the coregulator, in epithelial cells, and that MTA1 status is a nucleus to participate in the expression of TGF-b1-target determinant of TGF-b1-induced epithelial-to-mesenchymal genes (Deckers et al., 2006). Previous studies have shown transition (EMT) phenotypes. In addition, we found that that CUTL1, also known as CDP (CCAAT displacement MTA1/polymerase II/activator protein-1 (AP-1) co-activator protein), a target of TGF-b1, is needed for its short-term complex interacts with the FosB-gene chromatin and stimu- effects of TGF-b1 on cell motility involving Smad4- lates its transcription, and FosB in turn, utilizes FosB/histone dependent pathway (Michl et al.,2005). -

The Potential of the Natural Compound Neocarzilin a As Inhibitor of Cell Migration in Breast Cancer Cells
Dissertation zur Erlangung des Doktorgrades der Fakultät für Chemie und Pharmazie der Ludwig-Maximilians-Universität München The potential of the natural compound Neocarzilin A as inhibitor of cell migration in breast cancer cells Carolin Laura Pyka aus Düsseldorf Deutschland 2019 Erklärung Diese Dissertation wurde im Sinne von §7 der Promotionsordnung vom 28. November 2011 von Frau Prof. Dr. Angelika M. Vollmar betreut. Eidesstattliche Versicherung Diese Dissertation wurde eigenständig und ohne unerlaubte Hilfe erarbeitet. München, den 18.07.2019 Carolin Laura Pyka Dissertation eingereicht am: 18.07.2019 1. Gutachter: Prof. Dr. Angelika M. Vollmar 2. Gutachter: Prof. Dr. Johanna Pachmayr Mündliche Prüfung am: 28.08.2019 To my family CONTENTS Contents Contents Contents………………………………………………………………………………………………….I List of Figures………………………………………………………………………………………….IV List of Tables…………………………………………………………………………………………...V Abstract………………………………………………………………………………………………VIII 1 Introduction ....................................................................................................................... 1 1.1 Metastatic breast cancer – an overview ........................................................................................ 1 1.1.1 Facts and figures .................................................................................................................... 1 1.1.2 Classification of breast cancer ............................................................................................... 2 1.1.3 Treatment options for metastatic -

Micrornas in Cardiovascular Disease: from Pathogenesis to Prevention and Treatment
MicroRNAs in cardiovascular disease: from pathogenesis to prevention and treatment Daniel Quiat, Eric N. Olson J Clin Invest. 2013;123(1):11-18. https://doi.org/10.1172/JCI62876. Review Series The management of cardiovascular risk through lifestyle modification and pharmacotherapy is paramount to the prevention of cardiovascular disease. Epidemiological studies have identified obesity, dyslipidemia, diabetes, and hypertension as interrelated factors that negatively affect cardiovascular health. Recently, genetic and pharmacological evidence in model systems has implicated microRNAs as dynamic modifiers of disease pathogenesis. An expanded understanding of the function of microRNAs in gene regulatory networks associated with cardiovascular risk will enable identification of novel genetic mechanisms of disease and inform the development of innovative therapeutic strategies. Find the latest version: https://jci.me/62876/pdf Review series MicroRNAs in cardiovascular disease: from pathogenesis to prevention and treatment Daniel Quiat and Eric N. Olson Department of Molecular Biology, University of Texas Southwestern Medical Center, Dallas, Texas, USA. The management of cardiovascular risk through lifestyle modification and pharmacotherapy is paramount to the prevention of cardiovascular disease. Epidemiological studies have identified obesity, dyslipidemia, diabetes, and hypertension as interrelated factors that negatively affect cardiovascular health. Recently, genetic and pharmaco- logical evidence in model systems has implicated microRNAs -

Effects of the Pseudorabies Virus US3 Protein Kinase on Actin and Actin-Controlling Proteins
Universiteit Gent Faculteit Diergeneeskunde Vakgroep Virologie, Parasitologie en Immunologie Effects of the pseudorabies virus US3 protein kinase on actin and actin-controlling proteins Thary Jacob Proefschrift voorgedragen tot het behalen van de graad van Doctor in de Diergeneeskundige Wetenschappen, 2015 Promotor: Prof. dr. H. Favoreel ISBN: 9789058644459 © 2015 The author and the promoters give the authorization to consult and copy parts of this work for personal use only. Every other use is subject to copyright laws. The author should obtain written permission to reproduce any material contained in this work. Thary Jacob is supported by a personal PhD grant from the Agency for Innovation by Science and Technology in Flanders (IWT-Vlaanderen). This research was supported by a grant from the F.W.O.- Vlaanderen (Grant G.0835.09) and the Research Council of Ghent University (Concerted Research Action 01G01311). About the cover The front cover shows LifeAct (red) transduced swine testicle cells that were transfected with a plasmid encoding PRV US3. US3 protein was stained with primary mouse anti-US3 antibody and secondary FITC-labeled (green) anti-mouse antibody. Nuclei were counterstained with Hoechst 33342 (cyan). This image was taken using a Leica TCS SPE confocal microscope (XY length: 174.6 x 174.6 µm; Z = 6,8 µm, objective: ACS APO 63.0 x 1.30 (oil)). “What is written without effort is, in general, read without pleasure.” - Samuel Johnson Table of contents Abbreviation list ......................................................................................................................... -

PDF Hosted at the Radboud Repository of the Radboud University Nijmegen
PDF hosted at the Radboud Repository of the Radboud University Nijmegen The following full text is a publisher's version. For additional information about this publication click this link. http://hdl.handle.net/2066/147462 Please be advised that this information was generated on 2021-10-11 and may be subject to change. Chromatin Regulatory Complexes from an Interaction Proteomics Perspective an Interaction Proteomics from Regulatory Complexes Chromatin Chromatin Regulatory Complexes from an Interaction Proteomics Perspective H. İrem Baymaz H. İrem Baymaz H. İrem Baymaz 2015 Chromatin Regulatory Complexes from an Interaction Proteomics Perspective H. İrem Baymaz printed by: Proefschriftmaken.nl || Uitgeverij BOXPress Copyright © 2015 H. İrem Baymaz Chromatin Regulatory Complexes from an Interaction Proteomics Perspective Chromatine-Regulerende Complexen vanuit een Interactie-Proteomics Perspectief Proefschrift ter verkrijging van de graad van doctor aan de Radboud Universiteit Nijmegen op gezag van de rector magnificus prof. dr. Th.L.M. Engelen, volgens besluit van het college van decanen in het openbaar te verdedigen op donderdag 26 november 2015 om 12.30 uur precies door Hamdiye İrem Baymaz geboren op 8 maart 1986 te Ankara, Turkije Promoter: Prof. dr. M. Vermeulen Manuscriptcommissie: Prof. dr. J.H.L.M. van Bokhoven Prof. dr. H.T.M. Timmers (UMCU) Dr. L.M. Kamminga Anne ve Babama. Table of Contents List of Abbreviations 9 Chapter 1 11 Introduction Chapter 2 45 Identifying nuclear protein-protein interactions using GFP affinity purification -

Functional Analysis of the LIM Kinase 1 and Its Role in Cell Cycle Progression
Functional Analysis of the LIM kinase 1 and its role in Cell Cycle progression Inauguraldissertation zur Erlangung der Würde eines Doktors der Philosophie vorgelegt der Philosophisch-Naturwissenschaftlichen Fakultät der Universität Basel von Ayça Sayı aus Istanbul (Türkei) Zürich 2008 Genehmigt von der Philosophisch- Naturwissenschaftlichen Fakultät auf Antrag von Prof. Dr. Wilhelm Krek, Prof.Dr. Matthias Peter und Prof. Dr. Denis Monard. Basel, den 20. Juni 2006 Prof. Dr. Hans- Jakob Wirz Dekan Abstract LIMK1 (LIM-kinase 1) is a member of the LIMKs family of serine/threonine kinases, comprised of LIMK1, LIMK2 and testicular specific kinases, TESK1 and TESK2. These enzymes catalyze phosphorylation of an actin depolymerizing factor cofilin, thereby inactivate its depolymerizing activity, that leads to actin stabilization as filamentous actin (F-actin). As a consequence, LIMK1 plays a key role in actin cytoskeleton remodeling, notably in response to many extracellular cues that trigger the activation of the small Rho GTPases Rho, Rac and Cdc42, these latter being able to activate LIMK1 through their downstream effectors ROCK, PAK1 and PAK4 respectively. Moreover, on a pathological point of view, dysregulation of LIMK1 has being associated to several diseases: indeed, partial loss of LIMK1 is associated to a neurological disorder called William’s syndrome, whereas LIMK1 upregulation and/or hyperactivation is linked to cancer metastasis. The introduction chapter of this thesis details the current knowledge about LIMK1 and its func- tion in cell migration, cell cycle, neuronal differentiation, and phagocytosis. Additionally, the contribution of LIMK1 dysregulation in pathological circumstances, notably in WilliamÕs syn- drome and in cancer metastasis is discussed. The results chapter summarizes the work undertaken as partial fulfilment of this doctoral study. -

I3C & Hormone Positive Breast Cancer
ARTICLE IN PRESS Journal of Nutritional Biochemistry xx (2006) xxx–xxx Estrogen receptor a as a target for indole-3-carbinol Thomas T.Y. Wanga,4, Matthew J. Milnerb, John A. Milnerc, Young S. Kimc aPhytonutrients Laboratory, Beltsville Human Nutrition Research Center, Agricultural Research Service, U.S. Department of Agriculture, Beltsville, MD 20705, USA bDepartment of Biology, The Pennsylvania State University, University Park, PA, USA cNutritional Science Research Group, Division of Cancer Prevention, National Cancer Institute, National Institutes of Health, Bethesda, MD, USA Received 8 June 2005; received in revised form 14 October 2005; accepted 22 October 2005 Abstract A wealth of preclinical evidence supports the antitumorigenic properties of indole-3-carbinol (I3C), which is a major bioactive food component in cruciferous vegetables. However, the underlying molecular mechanism(s) accounting for these effects remain unresolved. In the present study, estrogen receptor alpha (ER-a) was identified as a potential molecular target for I3C. Treating MCF-7 cells with 100 AM I3C reduced ER-a mRNA expression by approximately 60% compared to controls. This reduction in ER-a transcript levels was confirmed using real-time polymerase chain reaction. The I3C dimer, 3,3V-diindolylmethane (DIM), was considerably more effective in depressing ER-a mRNA in MCF-7 cells than the monomeric unit. The suppressive effects of 5 AM DIM on ER-a mRNA was comparable to that caused by 100 AM I3C. DIM is known to accumulate in the nucleus and is a preferred ligand for aryl hydrocarbon receptor (AhR) to I3C. The addition of other AhR ligands, a-naphthoflavone (a-NF, 10 AM) and luteolin (10 AM), to the culture media resulted in a similar suppression in ER-a mRNA levels to that caused by 5 AM DIM. -
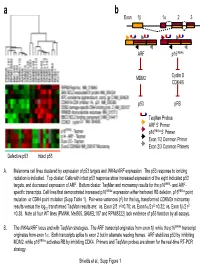
Taqman Probes ARF 5' Primer P16ink4a 5' Primer Exon 1/2
a b Exon 1β 1α 23 ARF p16INK4a MDM2 Cyclin D CDK4/6 p53 pRB TaqMan Probes ARF 5’ Primer p16INK4a 5’ Primer Exon 1/2 Common Primer Exon 2/3 Common Primers Defective p53 Intact p53 A. Melanoma cell lines clustered by expression of p53 targets and INK4a/ARF expression. The p53 response to ionizing radiation is indicated. Top cluster: Cells with intact p53 response show increased expression of the eight indicated p53 targets, and decreased expression of ARF. Bottom cluster: TaqMan and microarray results for the p16INK4- and ARF- specific transcripts. Cell lines that demonstrated increased p16INK4a expression either harbored RB deletion, p16INK4a point 2 mutation or CDK4 point mutation (Supp Table 1). Pair-wise variances (r ) for the log2 transformed CDKN2a microarray 2 2 2 results versus the log10 transformed TaqMan results are: vs. Exon 2/3 r =0.78; vs. Exon1α/2 r =0.32; vs. Exon 1β/2 r =0.38. Note all four WT lines (PMWK, Mel505, SKMEL187 and RPMI8322) lack evidence of p53 function by all assays. B. The INK4a/ARF locus and with TaqMan strategies. The ARF transcript originates from exon 1β while the p16INK4a transcript originates from exon 1α. Both transcripts splice to exon 2 but in alternate reading frames. ARF stabilizes p53 by inhibiting MDM2, while p16INK4a activates RB by inhibiting CDK4. Primers and TaqMan probes are shown for the real-time RT-PCR strategy. Shields et al., Supp Figure 1 SKMEL 28 U01 24h SKMEL WM2664 U01 48h WM2664 U01 24h 24 U01 48h SKMEL 24 U01 24h SKMEL 24 Untreated SKMEL 24 DMSO 48h SKMEL 24 DMSO 24h SKMEL WM2664 DMSO 24h WM2664 DMSO 48h WM2644 Untreated 28 Untreated SKMEL 28 DMSO 48h SKMEL 28 DMSO 24h SKMEL -3.00 -2.00 -1.00 0.00 1.00 2.00 3.00 relative to median expression Genes decreased by UO1 (863) HIF1A Hypoxia-inducible factor 1, alpha subunit NM_001530 RBBP8 Retinoblastoma binding protein 8 NM_002894 Homo sapiens, clone IMAGE:4337652, mRNA BC018676 EIF4EBP1 Eukaryotic translation initiation factor 4E binding protein EXOSC8 Exosome component 8 NM_181503 ENST00000321524 MCM7 MCM7 minichromosome maintenance deficient 7 (S. -

Microrna-125A-5P Targets LIM Kinase 1 to Inhibit Cisplatin Resistance of Cervical Cancer Cells
ONCOLOGY LETTERS 21: 392, 2021 MicroRNA-125a-5p targets LIM kinase 1 to inhibit cisplatin resistance of cervical cancer cells YONGQIAN XU*, YUJIE ZHENG*, YAN DUAN, LIN MA and PING NAN Department of Obstetrics and Gynecology, Shengli Oilfield Central Hospital, Dongying, Shandong 257000, P.R. China Received January 11, 2020; Accepted January 28, 2021 DOI: 10.3892/ol.2021.12653 Abstract. Cervical cancer (CC), also known as invasive miR‑125a‑5p could partially reverse the effect of LIMK1 on cervical carcinoma, is one of the most common gynecologic the proliferation, apoptosis, IC50 of DDP and the expressions malignancies. The aim of the present study was to investigate of drug resistance‑related proteins. The findings of the present the function of microRNA (miR)‑125a‑5p on CC progression study indicated that miR‑125a‑5p sensitizes CC cells to DDP and cisplatin (DDP) resistance. For this purpose, reverse by targeting LIMK1, hence increasing the anticancer efficacy transcription‑quantitative PCR (RT‑qPCR) was used to of cisplatin. assess the expression of miR‑125a‑5p and LIMK1 in CC tissues, corresponding normal tissues and cells (human Introduction CC cell lines: C‑33A, CaSKi; human cervical epithelial cells: HUCEC). Cisplatin (DDP) resistant cervical cancer Cervical carcinoma (CC) is one of the most common tumors cell lines were established (C‑33A/DDP and CaSKi/DDP among women in the world, and the incidence of which cell lines). RT‑qPCR results demonstrated that miR‑125a‑5p was ~7% in globally 2020 (1). In developed economies, the or LIM kinase 1 (LIMK1) expression was downregulated or 5‑year survival rate of patients with CC is >65%, whereas this upregulated in C‑33A/DDP and CaSKi/DDP cells, respec‑ proportion is <20% in developing countries (2). -

Transcriptional Mechanisms of WNT5A Based on NF-Κb, Hedgehog, Tgfß, and Notch Signaling Cascades
763-769 23/4/2009 01:21 ÌÌ Page 763 INTERNATIONAL JOURNAL OF MOLECULAR MEDICINE 23: 763-769, 2009 763 Transcriptional mechanisms of WNT5A based on NF-κB, Hedgehog, TGFß, and Notch signaling cascades MASUKO KATOH1 and MASARU KATOH2 1M&M Medical BioInformatics, Hongo 113-0033; 2Genetics and Cell Biology Section, National Cancer Center, Tokyo 104-0045, Japan Received March 5, 2009; Accepted April 2, 2009 DOI: 10.3892/ijmm_00000190 Abstract. WNT5A is a cancer-associated gene involved in were found to upregulate WNT5A expression directly through invasion and metastasis of melanoma, breast cancer, pancreatic the Smad complex, and also indirectly through Smad- cancer, and gastric cancer. WNT5A transduces signals through induced CUX1 and MAP3K7-mediated NF-κB. Together Frizzled, ROR1, ROR2 or RYK receptors to ß-catenin-TCF/ these facts indicate that WNT5A is transcribed based on LEF, DVL-RhoA-ROCK, DVL-RhoB-Rab4, DVL-Rac-JNK, multiple mechanisms, such as NF-κB, Hedgehog, TGFß, and DVL-aPKC, Calcineurin-NFAT, MAP3K7-NLK, MAP3K7- Notch signaling cascades. NF-κB, and DAG-PKC signaling cascades in a context- dependent manner. SNAI1 (Snail), CD44, G3BP2, and YAP1 Introduction are WNT5A target genes. We and other groups previously reported that IL6- or LIF-induced signaling through JAK- WNT signaling cascades are involved in a variety of cellular STAT3 signaling cascade is involved in WNT5A upregulation processes during embryogenesis and carcinogenesis (1-4). (STAT3-WNT5A signaling loop). Here, refined integrative Because biological functions of human genes and those of genomic analyses of WNT5A were carried out to elucidate model-animal orthologs are not always conserved due to other mechanisms of WNT5A transcription.