Identification of Genomic Loci Contributing to Agenesis of the Corpus Callosum Mary C
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Familial Intellectual Disability As a Result of a Derivative Chromosome
Zhang et al. Molecular Cytogenetics (2018) 11:18 DOI 10.1186/s13039-017-0349-x RESEARCH Open Access Familial intellectual disability as a result of a derivative chromosome 22 originating from a balanced translocation (3;22) in a four generation family Kaihui Zhang1†, Yan Huang2†, Rui Dong1†, Yali Yang2, Ying Wang1, Haiyan Zhang1, Yufeng Zhang1, Zhongtao Gai1* and Yi Liu1* Abstract Background: Balanced reciprocal translocation is usually an exchange of two terminal segments from different chromosomes without phenotypic effect on the carrier while leading to increased risk of generating unbalanced gametes. Here we describe a four-generation family in Shandong province of China with at least three patients sharing severe intellectual disability and developmental delay resulting from a derivative chromosome 22 originating from a balanced translocation (3;22) involving chromosomes 3q28q29 and 22q13.3. Methods: The proband and his relatives were detected by using karyotyping, chromosome microarray analysis, fluorescent in situ hybridization and real-time qPCR. Results: The proband, a 17 month-old boy, presented with severe intellectual disability, developmental delay, specific facial features and special posture of hands. Pedigree analysis showed that there were at least three affected patients. The proband and other two living patients manifested similar phenotypes and were identified to have identically abnormal cytogenetic result with an unbalanced translocation of 9.0 Mb duplication at 3q28q29 and a 1.7Mb microdeletion at 22q13.3 by karyotyping and chromosome microarray analysis. His father and other five relatives had a balanced translocation of 3q and 22q. Fluorescence in situ hybridization and real-time qPCR definitely validated the results. -

Genetic Causes.Pdf
1 September 2015 Genetic causes of childhood apraxia of speech: Case‐based introduction to DNA, inheritance, and clinical management Beate Peter, Ph.D., CCC‐SLP Assistant Professor Dpt. of Speech & Hearing Science Arizona State University Adjunct Assistant Professor AG Dpt. of Communication Sciences & Disorders ATAGCT Saint Louis University T TAGCT Affiliate Assistant Professor Dpt. of Speech & Hearing Sciences University of Washington 1 Disclosure Statement Disclosure Statement Dr. Peter is co‐editor of a textbook on speech development and disorders (B. Peter & A. MacLeod, Eds., 2013), for which she may receive royalty payments. If she shares information about her ongoing research study, this may result in referrals of potential research participants. She has no financial interest or related personal interest of bias in any organization whose products or services are described, reviewed, evaluated or compared in the presentation. 2 Agenda Topic Concepts Why we should care about genetics. Case 1: A sporadic case of CAS who is missing a • Cell, nucleus, chromosomes, genes gene. Introduction to the language of genetics • From genes to proteins • CAS can result when a piece of DNA is deleted or duplicated Case 2: A multigenerational family with CAS • How the FOXP2 gene was discovered and why research in genetics of speech and language disorders is challenging • Pathways from genes to proteins to brain/muscle to speech disorder Case 3: One family's quest for answers • Interprofessional teams, genetic counselors, medical geneticists, research institutes • Early signs of CAS, parent education, early intervention • What about genetic testing? Q&A 3 “Genetic Causes of CAS: Case-Based Introduction to DNA, Inheritance and Clinical Management,” Presented by: Beate Peter, PhD, CCC-SLP, September 29, 2015, Sponsored by: CASANA 2 Why should you care about genetics? 4 If you are a parent of a child with childhood apraxia of speech … 5 When she was in preschool, He doesn’t have any friends. -

3 Chromosome Chapter
Chromosome 3 ©Chromosome Disorder Outreach Inc. (CDO) Technical genetic content provided by Dr. Iosif Lurie, M.D. Ph.D Medical Geneticist and CDO Medical Consultant/Advisor. Ideogram courtesy of the University of Washington Department of Pathology: ©1994 David Adler.hum_03.gif Introduction The size of chromosome 3 is ~200 Mb. Within this chromosome, there are thousands of genes, many of which are necessary for normal intellectual development or involved in the formation of body organs. Deletions of Chromosome 3 The length of the short arm of chromosome 3 is ~90 Mb. Most known deletions of 3p are caused by the loss of its distal 15 Mb segment (3p25–pter). Deletions of the more proximal segments are relatively rare; there are only ~50 reports on such patients. Therefore, it would be premature to talk about any syndrome related to deletions of the proximal part of 3p. The location of the breakpoints, size of deletion, and reported abnormalities are different in most described patients. However, recurrent aortal stenosis in patients with deletion 3p11p14.2, abnormal lung lobation in patients with deletion 3p12p14.2, agenesis or hypoplasia of the corpus callosum in patients with deletion 3p13, microphthalmia and coloboma in patients with deletion 3p13p21.1, choanal atresia and absent gallbladder in patients with deletion 3p13p21, and hearing loss in patients with deletion 3p14 are all indicators that the above–mentioned segments likely contain genes involved in the formation of these systems. Deletions of 3p Deletion of 3p25–pter The most distal segment of the short arm of chromosome 3 is 3p26 and spans ~8 Mb. -

14Q13 Deletions FTNW
14q13 deletions rarechromo.org 14q13 deletions A chromosome 14 deletion means that part of one of the body’s chromosomes (chromosome 14) has been lost or deleted. If the deleted material contains important genes, learning disability, developmental delay and health problems may occur. How serious these problems are depends on how much of the chromosome has been deleted, which genes have been lost and where precisely the deletion is. The features associated with 14q13 deletions vary from person to person, but are likely to include a degree of developmental delay, an unusually small or large head, a raised risk of medical problems and unusual facial features. Genes and chromosomes Our bodies are made up of billions of cells. Most of these cells contain a complete set of thousands of genes that act as instructions, controlling our growth, development and how our bodies work. Inside human cells there is a nucleus where the genes are carried on microscopically small, thread-like structures called chromosomes which are made up p arm p arm of DNA. p arm p arm Chromosomes come in pairs of different sizes and are numbered from largest to smallest, roughly according to their size, from number 1 to number 22. In addition to these so-called autosomal chromosomes there are the sex chromosomes, X and Y. So a human cell has 46 chromosomes: 23 inherited from the mother and 23 inherited from the father, making two sets of 23 chromosomes. A girl has two X chromosomes (XX) while a boy will have one X and one Y chromosome (XY). -

Inside This Issue
Winter 2014 No. 77 Inside this issue Group News | Fundraising | Members’ Letters | One Family Living with Two Different Chromosome Disorders | Bristol Conference 2014 | Unique Leaflets | Christmas Card Order Form Sophie, Unique’s Chair of Trustees Dear Members, In the past month a few things have reminded me of why it is so important to make connections through Unique but also to draw support from other parents around us. I’ve just returned from Unique’s most recent family conference in Bristol where 150 of us parents and carers had a lovely time in workshops, meals and activities, chatting and watching our children milling around together like one big family since – although we had never met before – we have shared so many experiences in common. However in contrast I have also just met a new mum who has just moved to my area from far away with two toddlers, one with a rare joys of the internet, it is becoming easier to meet others with similar, chromosome disorder, who is starting from scratch with no even very rare, chromosome disorders around the world and to find professional, medical or social support. She reminds me of how yourself talking to them in the middle of the night about some lonely I felt when Max was newly diagnosed, when I knew no one interesting things our children share in common (obsession with with a disabled child let alone anyone with a rare chromosome catalogues, anyone?) And of course we also have an enormous disorder. Elsewhere our latest Unique Facebook group, Unique amount in common with so many parents of children with other Russia, is also just starting up – so far it includes just a small special needs or disabilities around us in our own communities who number of members sharing very different experiences to mine here will often be walking the same path as us. -

©Ferrata Storti Foundation
Haematologica 2000; 85:1207-1210 Acute Leukemias case of the month Cytogenetic characterization of acute myeloid leukemia in Shwachman's syndrome. A case report FRANCESCA R. SPIRITO, BARBARA CRESCENZI, CATERINA MATTEUCCI, MASSIMO F. MARTELLI, CRISTINA MECUCCI Hematology and Bone Marrow Transplantation Unit, University of Perugia, Italy ABSTRACT We report on a case of acute myeloid leukemia in a tion. A recent analysis of 21 patients with SDS 17-year old boy affected by Shwachman Diamond showed that the incidence of myelodysplasia syndrome (SDS). Conventional cytogenetics at diag- and transformation to acute myeloid leukemia nosis revealed an abnormal clone with complex 4 karyotypic changes including typical myeloid aber- is 33% and 24%, respectively. A few cases of rations, such as monosomy 5, tetrasomy of chro- acute lymphoblastic leukemia have also been 5-7 mosome 8, trisomy 9, and deletion of the short arm described as well as one case of a juvenile form of chromosome 12. The boy was treated with con- of chronic myeloid leukemia.8 When leukemia ventional chemotherapy and reached complete develops, due to the preceding bone marrow remission of leukemia, confirmed by cytogenetics failure, serious complications may be induced and fluorescence in situ hybridization. Nevertheless by chemotherapy and, because of the underly- he failed to regenerate normal marrow cellularity ing organ dysfunctions, these patients should and blood cell count. Cytogenetic information on be managed with extreme caution when treat- hematologic malignancies in SDS patients are dis- cussed. ed with allogeneic bone marrow transplanta- 9-13 ©2000; Ferrata Storti Foundation tion. We describe here the cytogenetic char- acterization and clinical course of an acute Key words: Shwachman’s syndrome, leukemia, cytogenetics myeloid leukemia occurring in a young patient affected by Shwachman's syndrome. -
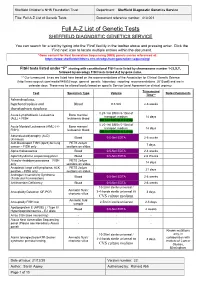
Full A-Z List of Genetic Tests Document Reference Number: 413.001
Sheffield Children’s NHS Foundation Trust Department: Sheffield Diagnostic Genetics Service Title: Full A-Z List of Genetic Tests Document reference number: 413.001 Full A-Z List of Genetic Tests SHEFFIELD DIAGNOSTIC GENETICS SERVICE You can search for a test by typing into the ‘Find’ facility in the toolbar above and pressing enter. Click the ‘Find next’ icon to locate multiple entries within the document. *Gene content for Next Generation Sequencing (NGS) panels can be referenced at: https://www.sheffieldchildrens.nhs.uk/sdgs/next-generation-sequencing/ FISH tests listed under “F” starting with constitutional FISH tests listed by chromosome number 1-22,X,Y, followed by oncology FISH tests listed A-Z by gene name. ** Our turnaround times are listed here based on the recommendations of the Association for Clinical Genetic Science (http://www.acgs.uk.com/media/949852/acgs_general_genetic_laboratory_reporting_recommendations_2015.pdf) and are in calendar days. These may be altered locally based on specific Service Level Agreement or clinical urgency. Turnaround Test Specimen Type Volume Notes/Comments Time** Achondroplasia, hypchondroplasia and Blood 0.5-5ml 2-6 weeks thanatophoric dysplasia 0.25-1ml BM in 5-10ml of Acute Lymphoblastic Leukaemia Bone marrow/ transport medium 14 days (ALL) + FISH leukaemic blood OR 1ml BM/VB in Li Hep 0.25-1ml BM in 5-10ml of Acute Myeloid Leukaemia (AML) (+/- Bone marrow/ transport medium 14 days FISH) leukaemic blood OR 1ml BM/VB in Li Hep Adrenoleukodystrophy (ALD) Blood 0.5-5ml EDTA 2-6 weeks (X-linked) -

Role and Regulation of the P53-Homolog P73 in the Transformation of Normal Human Fibroblasts
Role and regulation of the p53-homolog p73 in the transformation of normal human fibroblasts Dissertation zur Erlangung des naturwissenschaftlichen Doktorgrades der Bayerischen Julius-Maximilians-Universität Würzburg vorgelegt von Lars Hofmann aus Aschaffenburg Würzburg 2007 Eingereicht am Mitglieder der Promotionskommission: Vorsitzender: Prof. Dr. Dr. Martin J. Müller Gutachter: Prof. Dr. Michael P. Schön Gutachter : Prof. Dr. Georg Krohne Tag des Promotionskolloquiums: Doktorurkunde ausgehändigt am Erklärung Hiermit erkläre ich, dass ich die vorliegende Arbeit selbständig angefertigt und keine anderen als die angegebenen Hilfsmittel und Quellen verwendet habe. Diese Arbeit wurde weder in gleicher noch in ähnlicher Form in einem anderen Prüfungsverfahren vorgelegt. Ich habe früher, außer den mit dem Zulassungsgesuch urkundlichen Graden, keine weiteren akademischen Grade erworben und zu erwerben gesucht. Würzburg, Lars Hofmann Content SUMMARY ................................................................................................................ IV ZUSAMMENFASSUNG ............................................................................................. V 1. INTRODUCTION ................................................................................................. 1 1.1. Molecular basics of cancer .......................................................................................... 1 1.2. Early research on tumorigenesis ................................................................................. 3 1.3. Developing -

See Shalini C Reshmi's Curriculum Vitae
Shalini C. Reshmi, Ph.D. Contact Information: Institute for Genomic Medicine Nationwide Children’s Hospital 575 Childrens Crossroads, RB3-WB2233 Columbus, OH 43215 Lab: (614) 722-5321 Fax: (614) 355-6833 [email protected] CURRENT ACADEMIC POSITIONS 2/2018-present Assistant Professor, Department of Pediatrics, The Ohio State University, Columbus, OH 7/2009-present Assistant Professor (Clinical), Department of Pathology, The Ohio State University, Columbus, OH ADMINISTRATIVE APPOINTMENT 10/2016-present Director, Clinical Laboratory Institute for Genomic Medicine Nationwide Children's Hospital, Columbus, OH 9/2012-10/2016 Associate Director, Cytogenetics/Molecular Genetics Laboratory Nationwide Children's Hospital, Columbus, OH 12/2008-9/2012 Assistant Director, Cytogenetics/Molecular Genetics Laboratory Nationwide Children's Hospital, Columbus, OH EDUCATION 9/2001-5/2005 Ph.D. Human Genetics University of Pittsburgh, Graduate School of Public Health Pittsburgh, PA 9/1994-4/1996 M.S. Human Genetics University of Pittsburgh, Graduate School of Public Health Pittsburgh, PA 8/1988-5/1992 B.S. Biology The Catholic University of America Washington, D.C. TRAINING 7/2007-10/2008 American College of Medical Genetics and Genomics Clinical Molecular Genetics University of Chicago, Pritzker School of Medicine Chicago, IL 7/2005-6/2007 American College of Medical Genetics and Genomics Clinical Cytogenetics University of Chicago, Pritzker School of Medicine Chicago, IL Board Certifications 2 9/01/09- Diplomate, American Board of -

The Cytogenetics of Hematologic Neoplasms 1 5
The Cytogenetics of Hematologic Neoplasms 1 5 Aurelia Meloni-Ehrig that errors during cell division were the basis for neoplastic Introduction growth was most likely the determining factor that inspired early researchers to take a better look at the genetics of the The knowledge that cancer is a malignant form of uncon- cell itself. Thus, the need to have cell preparations good trolled growth has existed for over a century. Several biologi- enough to be able to understand the mechanism of cell cal, chemical, and physical agents have been implicated in division became of critical importance. cancer causation. However, the mechanisms responsible for About 50 years after Boveri’s chromosome theory, the this uninhibited proliferation, following the initial insult(s), fi rst manuscripts on the chromosome makeup in normal are still object of intense investigation. human cells and in genetic disorders started to appear, fol- The fi rst documented studies of cancer were performed lowed by those describing chromosome changes in neoplas- over a century ago on domestic animals. At that time, the tic cells. A milestone of this investigation occurred in 1960 lack of both theoretical and technological knowledge with the publication of the fi rst article by Nowell and impaired the formulations of conclusions about cancer, other Hungerford on the association of chronic myelogenous leu- than the visible presence of new growth, thus the term neo- kemia with a small size chromosome, known today as the plasm (from the Greek neo = new and plasma = growth). In Philadelphia (Ph) chromosome, to honor the city where it the early 1900s, the fundamental role of chromosomes in was discovered (see also Chap. -

Bone Marrow Karyotypes of Children with Nonlymphocytic Leukemia
Pediat. Res. 13: 1247-1254 (1979) Chromosome abnormalities nonlymphotic leukemia leukemia, childhood Bone Marrow Karyotypes of Children with Nonlymphocytic Leukemia A. HAGEMEIJER, G. E. VAN ZANEN, E. M. E. SMIT, AND K. HAHLEN Department of Cell Biology and Genetics (A. H.. E. M.E. S.) and Department of Pediatric Oncology (G. E. K Z.. K H.) Sophia Children's Hospital, Erasmus University, Rotterdam, The Netherlands Summary studies with modern banding techniques. The Ph' chromosome may be found in some cases if childhbod CML, and its presence Bone marrow (BM) karyotypes from 16 consecutive children can differentiate the adult type of CML with its more protracted presenting with nonlymphocytic leukemia were established with course, from the juvenile variant. Nonrandom acquired abnor- the use of banding techniques, before therapy. The two patients malities have also been reported in a small number of children with chronic myeloid leukemia (CML) showed the Philadelphia with ANLL: monosomy 7 was reported three times in AML, (Phl) translocation (9q+;22q-). Five of the 14 patients with an AMMoL, and EL (7, 10, 17) deletion 7q22 was found twice in acute nonlymphocytic leukemia (ANLL) presented no acquired AML (9), and monosomy 9 was described once in AML (8). An cytogenetic abnormalities, but one of these five showed a high extensive report of cytogenetic abnormalities in childhood leuke- level of hypodiploidy. One patient with AML evidenced a variant mia has been published by Zuelzer et al. in 1976 (3 l), but it deals of the Phl chromosome originated as a translocation (12p+;22q-). mainly with acute lymphocytic leukemia; furthermore, this study Nonrandom abnormalities (-7; 7q-; +8; t(8;21); -21) were found started before the use of banding techniques, making the karyo- in six patients, isolated or in association with other aberrations. -

Cytogenetic Abnormalities in 772 ... -.: Scientific Press International Limited
Journal of Applied Medical Sciences, vol. 3, no. 3, 2014, 19-29 ISSN: 2241-2328 (print version), 2241-2336 (online) Scienpress Ltd, 2014 Cytogenetic Abnormalities in 772 Patients with Mental Retardation (MR) in Southern Region of Turkey: Report and Review Osman Demirhan1*, Nilgün Tanrıverdi1, Erdal Tunç1, Dilara Süleymanova1 and Deniz Taştemir2 Abstract Mental retardation (MR) is a heterogeneous condition, affecting 1-3% of general population. Identification of chromosomal abnormalities (CAs) associated with mental disorders may be especially important given the unknown pathophysiology and the probable genetic heterogeneity of MR. In this study, we aimed to evaluate karyotype results of 772 MR cases, retrospectively. For karyotyping, standard lymphocyte culturing and GTG banding methods were done. For analysis, cytovision software was used. In result, out of 772 MR cases, 87 cases showed abnormal chromosomal constitutions (11.3%), and normal karyotype results were detected in 88.7% of all patients. Numerical and structural CAs were detected in 2.6% (20 of 772) and 8.7 (67 of 772) of cases, respectively. This study revealed Down syndrome as the most common chromosomal abnormality (1%). The ratio of X chromosome monosomy was 0.6%. In conclusion, patients with MR should be routinely karyotyped. Interesting CAs we found, may harbor important genes for MR and give important tips for linkage in possible genome scan projects. Keywords: Chromosomal abnormalities, GTG-banding, karyotype, MR 1 Introduction MR is the most frequent cause of serious handicap in children and young adults with an estimated prevalence up to 1-3% of the population, and is one of the more important topics in medical science [1,2].