Bacterial Community Assemblages in Classroom Floor Dust of 50 Public
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Bacillus Crassostreae Sp. Nov., Isolated from an Oyster (Crassostrea Hongkongensis)
International Journal of Systematic and Evolutionary Microbiology (2015), 65, 1561–1566 DOI 10.1099/ijs.0.000139 Bacillus crassostreae sp. nov., isolated from an oyster (Crassostrea hongkongensis) Jin-Hua Chen,1,2 Xiang-Rong Tian,2 Ying Ruan,1 Ling-Ling Yang,3 Ze-Qiang He,2 Shu-Kun Tang,3 Wen-Jun Li,3 Huazhong Shi4 and Yi-Guang Chen2 Correspondence 1Pre-National Laboratory for Crop Germplasm Innovation and Resource Utilization, Yi-Guang Chen Hunan Agricultural University, 410128 Changsha, PR China [email protected] 2College of Biology and Environmental Sciences, Jishou University, 416000 Jishou, PR China 3The Key Laboratory for Microbial Resources of the Ministry of Education, Yunnan Institute of Microbiology, Yunnan University, 650091 Kunming, PR China 4Department of Chemistry and Biochemistry, Texas Tech University, Lubbock, TX 79409, USA A novel Gram-stain-positive, motile, catalase- and oxidase-positive, endospore-forming, facultatively anaerobic rod, designated strain JSM 100118T, was isolated from an oyster (Crassostrea hongkongensis) collected from the tidal flat of Naozhou Island in the South China Sea. Strain JSM 100118T was able to grow with 0–13 % (w/v) NaCl (optimum 2–5 %), at pH 5.5–10.0 (optimum pH 7.5) and at 5–50 6C (optimum 30–35 6C). The cell-wall peptidoglycan contained meso-diaminopimelic acid as the diagnostic diamino acid. The predominant respiratory quinone was menaquinone-7 and the major cellular fatty acids were anteiso-C15 : 0, iso-C15 : 0,C16 : 0 and C16 : 1v11c. The polar lipids consisted of diphosphatidylglycerol, phosphatidylethanolamine, phosphatidylglycerol, an unknown glycolipid and an unknown phospholipid. The genomic DNA G+C content was 35.9 mol%. -

Bacterial Communities of the Upper Respiratory Tract of Turkeys
www.nature.com/scientificreports OPEN Bacterial communities of the upper respiratory tract of turkeys Olimpia Kursa1*, Grzegorz Tomczyk1, Anna Sawicka‑Durkalec1, Aleksandra Giza2 & Magdalena Słomiany‑Szwarc2 The respiratory tracts of turkeys play important roles in the overall health and performance of the birds. Understanding the bacterial communities present in the respiratory tracts of turkeys can be helpful to better understand the interactions between commensal or symbiotic microorganisms and other pathogenic bacteria or viral infections. The aim of this study was the characterization of the bacterial communities of upper respiratory tracks in commercial turkeys using NGS sequencing by the amplifcation of 16S rRNA gene with primers designed for hypervariable regions V3 and V4 (MiSeq, Illumina). From 10 phyla identifed in upper respiratory tract in turkeys, the most dominated phyla were Firmicutes and Proteobacteria. Diferences in composition of bacterial diversity were found at the family and genus level. At the genus level, the turkey sequences present in respiratory tract represent 144 established bacteria. Several respiratory pathogens that contribute to the development of infections in the respiratory system of birds were identifed, including the presence of Ornithobacterium and Mycoplasma OTUs. These results obtained in this study supply information about bacterial composition and diversity of the turkey upper respiratory tract. Knowledge about bacteria present in the respiratory tract and the roles they can play in infections can be useful in controlling, diagnosing and treating commercial turkey focks. Next-generation sequencing has resulted in a marked increase in culture-independent studies characterizing the microbiome of humans and animals1–6. Much of these works have been focused on the gut microbiome of humans and other production animals 7–11. -

Xiexin Tang Improves the Symptom of Type 2 Diabetic Rats by Modulation of the Gut Microbiota
www.nature.com/scientificreports OPEN Xiexin Tang improves the symptom of type 2 diabetic rats by modulation of the gut microbiota Received: 30 August 2017 Xiaoyan Wei, Jinhua Tao , Suwei Xiao, Shu Jiang, Erxin Shang, Zhenhua Zhu, Dawei Qian Accepted: 13 February 2018 & Jinao Duan Published: xx xx xxxx Type 2 diabetes mellitus (T2DM), a chronic metabolic disease which severely impairs peoples’ quality of life, currently attracted worldwide concerns. There are growing evidences that gut microbiota can exert a great impact on the development of T2DM. Xiexin Tang (XXT), a traditional Chinese medicine prescription, has been clinically used to treat diabetes for thousands of years. However, few researches are investigated on the modulation of gut microbiota community by XXT which will be very helpful to unravel how it works. In this study, bacterial communities were analyzed based on high-throughput 16S rRNA gene sequencing. Results indicated that XXT could notably shape the gut microbiota. T2DM rats treated with XXT exhibited obvious changes in the composition of the gut microbiota, especially for some short chain fatty acids producing and anti-infammatory bacteria such as Adlercreutzia, Alloprevotella, Barnesiella, [Eubacterium] Ventriosum group, Blautia, Lachnospiraceae UCG-001, Papillibacter and Prevotellaceae NK3B31 group. Additionally, XXT could also signifcantly ameliorate hyperglycemia, lipid metabolism dysfunction and infammation in T2DM rats. Moreover, the correlation analysis illustrated that the key microbiota had a close relationship with the T2DM related indexes. The results probably provided useful information for further investigation on its active mechanism and clinical application. T2DM, a chronic metabolic disease characterized by hyperglycemia as a result of insufcient insulin secretion, insulin action or both1, is estimated that its numbers in the adults will increase by 55% by 20352. -

Product Sheet Info
Product Information Sheet for HM-289 Facklamia sp., Strain HGF4 Incubation: Temperature: 37°C Catalog No. HM-289 Atmosphere: Aerobic Propagation: 1. Keep vial frozen until ready for use, then thaw. For research use only. Not for human use. 2. Transfer the entire thawed aliquot into a single tube of broth. Contributor: 3. Use several drops of the suspension to inoculate an Thomas M. Schmidt, Professor, Department of Microbiology agar slant and/or plate. and Molecular Genetics, Michigan State University, East 4. Incubate the tube, slant and/or plate at 37°C for 48 to Lansing, Michigan, USA 168 hours. Manufacturer: Citation: BEI Resources Acknowledgment for publications should read “The following reagent was obtained through BEI Resources, NIAID, NIH as Product Description: part of the Human Microbiome Project: Facklamia sp., Strain Bacteria Classification: Aerococcaceae, Facklamia HGF4, HM-289.” Species: Facklamia sp. Strain: HGF4 Biosafety Level: 2 Original Source: Facklamia sp., strain HGF4 is a human Appropriate safety procedures should always be used with gastrointestinal isolate.1 this material. Laboratory safety is discussed in the following Comments: Facklamia sp., strain HGF4 (HMP ID 9411) is a publication: U.S. Department of Health and Human Services, reference genome for The Human Microbiome Project Public Health Service, Centers for Disease Control and (HMP). HMP is an initiative to identify and characterize Prevention, and National Institutes of Health. Biosafety in human microbial flora. The complete genome of Facklamia Microbiological and Biomedical Laboratories. 5th ed. sp., strain HGF4 is currently being sequenced at the J. Washington, DC: U.S. Government Printing Office, 2009; see Craig Venter Institute. -

Gut Microbiome Composition Remains Stable in Individuals with Diabetes-Related Early to Late Stage Chronic Kidney Disease
biomedicines Article Gut Microbiome Composition Remains Stable in Individuals with Diabetes-Related Early to Late Stage Chronic Kidney Disease Ashani Lecamwasam 1,2,3,*, Tiffanie M. Nelson 4, Leni Rivera 3, Elif I. Ekinci 2,5, Richard Saffery 1,6 and Karen M. Dwyer 3 1 Epigenetics Research, Murdoch Children’s Research Institute, VIC 3052, Australia; [email protected] 2 Department of Endocrinology, Austin Health, VIC 3079, Australia; [email protected] 3 School of Medicine, Faculty of Health, Deakin University, VIC 3220, Australia; [email protected] (L.R.); [email protected] (K.M.D.) 4 Menzies Health Institute Queensland, Griffith University, QLD 4222, Australia; [email protected] 5 Department of Medicine, University of Melbourne, VIC 3010, Australia 6 Department of Paediatrics, University of Melbourne, VIC 3010, Australia * Correspondence: [email protected]; Tel.: +613-8341-6200; Fax: +613-9348-1391 Abstract: (1) Background: Individuals with diabetes and chronic kidney disease display gut dysbiosis when compared to healthy controls. However, it is unknown whether there is a change in dysbiosis across the stages of diabetic chronic kidney disease. We investigated a cross-sectional study of patients with early and late diabetes associated chronic kidney disease to identify possible microbial differences between these two groups and across each of the stages of diabetic chronic kidney disease. (2) Methods: This cross-sectional study recruited 95 adults. DNA extracted from collected stool samples were used for 16S rRNA sequencing to identify the bacterial community in the Citation: Lecamwasam, A.; Nelson, gut. (3) Results: The phylum Firmicutes was the most abundant and its mean relative abundance T.M.; Rivera, L.; Ekinci, E.I.; Saffery, was similar in the early and late chronic kidney disease group, 45.99 ± 0.58% and 49.39 ± 0.55%, R.; Dwyer, K.M. -
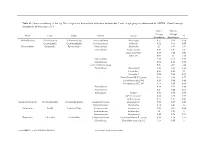
Genus Contributing to the Top 70% of Significant Dissimilarity of Bacteria Between Day 7 and 18 Age Groups As Determined by SIMPER
Table S1: Genus contributing to the top 70% of significant dissimilarity of bacteria between day 7 and 18 age groups as determined by SIMPER. Overall average dissimilarity between ages is 51%. Day 7 Day 18 Average Average Phyla Class Order Family Genus % Abundance Abundance Actinobacteria Actinobacteria Actinomycetales Actinomycetaceae Actinomyces 0.57 0.24 0.74 Coriobacteriia Coriobacteriales Coriobacteriaceae Collinsella 0.51 0.61 0.57 Bacteroidetes Bacteroidia Bacteroidales Bacteroidaceae Bacteroides 2.1 1.63 1.03 Marinifilaceae Butyricimonas 0.82 0.81 0.9 Sanguibacteroides 0.08 0.42 0.59 CAG-873 0.01 1.1 1.68 Marinifilaceae 0.38 0.78 0.91 Marinifilaceae 0.78 1.04 0.97 p-2534-18B5 gut group 0.06 0.7 1.04 Prevotellaceae Alloprevotella 0.46 0.83 0.89 Prevotella 2 0.95 1.11 1.3 Prevotella 7 0.22 0.42 0.56 Prevotellaceae NK3B31 group 0.62 0.68 0.77 Prevotellaceae UCG-003 0.26 0.64 0.89 Prevotellaceae UCG-004 0.11 0.48 0.68 Prevotellaceae 0.64 0.52 0.89 Prevotellaceae 0.27 0.42 0.55 Rikenellaceae Alistipes 0.39 0.62 0.77 dgA-11 gut group 0.04 0.38 0.56 RC9 gut group 0.79 1.17 0.95 Epsilonbacteraeota Campylobacteria Campylobacterales Campylobacteraceae Campylobacter 0.58 0.83 0.97 Helicobacteraceae Helicobacter 0.12 0.41 0.6 Firmicutes Bacilli Lactobacillales Enterococcaceae Enterococcus 0.36 0.31 0.64 Lactobacillaceae Lactobacillus 1.4 1.24 1 Streptococcaceae Streptococcus 0.82 0.58 0.53 Firmicutes Clostridia Clostridiales Christensenellaceae Christensenellaceae R-7 group 0.38 1.36 1.56 Clostridiaceae Clostridium sensu stricto 1 1.16 0.58 -

Role of Actinobacteria and Coriobacteriia in the Antidepressant Effects of Ketamine in an Inflammation Model of Depression
Pharmacology, Biochemistry and Behavior 176 (2019) 93–100 Contents lists available at ScienceDirect Pharmacology, Biochemistry and Behavior journal homepage: www.elsevier.com/locate/pharmbiochembeh Role of Actinobacteria and Coriobacteriia in the antidepressant effects of ketamine in an inflammation model of depression T Niannian Huanga,1, Dongyu Huaa,1, Gaofeng Zhana, Shan Lia, Bin Zhub, Riyue Jiangb, Ling Yangb, ⁎ ⁎ Jiangjiang Bia, Hui Xua, Kenji Hashimotoc, Ailin Luoa, , Chun Yanga, a Department of Anesthesiology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, China b Department of Internal Medicine, The Third Affiliated Hospital of Soochow University, Changzhou 213003, China c Division of Clinical Neuroscience, Chiba University Center for Forensic Mental Health, Chiba 260-8670, Japan ARTICLE INFO ABSTRACT Keywords: Ketamine, an N-methyl-D-aspartic acid receptor (NMDAR) antagonist, elicits rapid-acting and sustained anti- Ketamine depressant effects in treatment-resistant depressed patients. Accumulating evidence suggests that gut microbiota Depression via the gut-brain axis play a role in the pathogenesis of depression, thereby contributing to the antidepressant Lipopolysaccharide actions of certain compounds. Here we investigated the role of gut microbiota in the antidepressant effects of Gut microbiota ketamine in lipopolysaccharide (LPS)-induced inflammation model of depression. Ketamine (10 mg/kg) sig- nificantly attenuated the increased immobility time in forced swimming test (FST), which was associated with the improvements in α-diversity, consisting of Shannon, Simpson and Chao 1 indices. In addition to α-diversity, β-diversity, such as principal coordinates analysis (PCoA), and linear discriminant analysis (LDA) coupled with effect size measurements (LEfSe), showed a differential profile after ketamine treatment. -

Demonstrating the Potential of Abiotic Stress-Tolerant Jeotgalicoccus Huakuii NBRI 13E for Plant Growth Promotion and Salt Stress Amelioration
Annals of Microbiology (2019) 69:419–434 https://doi.org/10.1007/s13213-018-1428-x ORIGINAL ARTICLE Demonstrating the potential of abiotic stress-tolerant Jeotgalicoccus huakuii NBRI 13E for plant growth promotion and salt stress amelioration Sankalp Misra1,2 & Vijay Kant Dixit 1 & Shashank Kumar Mishra1,2 & Puneet Singh Chauhan1,2 Received: 10 September 2018 /Accepted: 20 December 2018 /Published online: 2 January 2019 # Università degli studi di Milano 2019 Abstract The present study aimed to demonstrate the potential of abiotic stress-tolerant Jeotgalicoccus huakuii NBRI 13E for plant growth promotion and salt stress amelioration. NBRI 13E was characterized for abiotic stress tolerance and plant growth-promoting (PGP) attributes under normal and salt stress conditions. Phylogenetic comparison of NBRI 13E was carried out with known species of the same genera based on 16S rRNA gene. Plant growth promotion and rhizosphere colonization studies were determined under greenhouse conditions using maize, tomato, and okra. Field experiment was also performed to assess the ability of NBRI 13E inoculation for improving growth and yield of maize crop in alkaline soil. NBRI 13E demonstrated abiotic stress tolerance and different PGP attributes under in vitro conditions. Phylogenetic and differential physiological analysis revealed considerable differences in NBRI 13E as compared with the reported species for Jeotgalicoccus genus. NBRI 13E colonizes in the rhizosphere of the tested crops, enhances plant growth, and ameliorates salt stress in a greenhouse experiment. Modulation in defense enzymes, chlorophyll, proline, and soluble sugar content in NBRI 13E-inoculated plants leads to mitigate the deleterious effect of salt stress. Furthermore, field evaluation of NBRI 13E inoculation using maize was carried out with recommended 50 and 100% chemical fertilizer controls, which resulted in significant enhancement of all vegetative parameters and total yield as compared to respective controls. -

Gut Microbiota Differs in Composition and Functionality Between Children
Diabetes Care Volume 41, November 2018 2385 Gut Microbiota Differs in Isabel Leiva-Gea,1 Lidia Sanchez-Alcoholado,´ 2 Composition and Functionality Beatriz Mart´ın-Tejedor,1 Daniel Castellano-Castillo,2,3 Between Children With Type 1 Isabel Moreno-Indias,2,3 Antonio Urda-Cardona,1 Diabetes and MODY2 and Healthy Francisco J. Tinahones,2,3 Jose´ Carlos Fernandez-Garc´ ´ıa,2,3 and Control Subjects: A Case-Control Mar´ıa Isabel Queipo-Ortuno~ 2,3 Study Diabetes Care 2018;41:2385–2395 | https://doi.org/10.2337/dc18-0253 OBJECTIVE Type 1 diabetes is associated with compositional differences in gut microbiota. To date, no microbiome studies have been performed in maturity-onset diabetes of the young 2 (MODY2), a monogenic cause of diabetes. Gut microbiota of type 1 diabetes, MODY2, and healthy control subjects was compared. PATHOPHYSIOLOGY/COMPLICATIONS RESEARCH DESIGN AND METHODS This was a case-control study in 15 children with type 1 diabetes, 15 children with MODY2, and 13 healthy children. Metabolic control and potential factors mod- ifying gut microbiota were controlled. Microbiome composition was determined by 16S rRNA pyrosequencing. 1Pediatric Endocrinology, Hospital Materno- Infantil, Malaga,´ Spain RESULTS 2Clinical Management Unit of Endocrinology and Compared with healthy control subjects, type 1 diabetes was associated with a Nutrition, Laboratory of the Biomedical Research significantly lower microbiota diversity, a significantly higher relative abundance of Institute of Malaga,´ Virgen de la Victoria Uni- Bacteroides Ruminococcus Veillonella Blautia Streptococcus versityHospital,Universidad de Malaga,M´ alaga,´ , , , , and genera, and a Spain lower relative abundance of Bifidobacterium, Roseburia, Faecalibacterium, and 3Centro de Investigacion´ BiomedicaenRed(CIBER)´ Lachnospira. -

The Microbiota Continuum Along the Female Reproductive Tract and Its Relation to Uterine-Related Diseases
ARTICLE DOI: 10.1038/s41467-017-00901-0 OPEN The microbiota continuum along the female reproductive tract and its relation to uterine-related diseases Chen Chen1,2, Xiaolei Song1,3, Weixia Wei4,5, Huanzi Zhong 1,2,6, Juanjuan Dai4,5, Zhou Lan1, Fei Li1,2,3, Xinlei Yu1,2, Qiang Feng1,7, Zirong Wang1, Hailiang Xie1, Xiaomin Chen1, Chunwei Zeng1, Bo Wen1,2, Liping Zeng4,5, Hui Du4,5, Huiru Tang4,5, Changlu Xu1,8, Yan Xia1,3, Huihua Xia1,2,9, Huanming Yang1,10, Jian Wang1,10, Jun Wang1,11, Lise Madsen 1,6,12, Susanne Brix 13, Karsten Kristiansen1,6, Xun Xu1,2, Junhua Li 1,2,9,14, Ruifang Wu4,5 & Huijue Jia 1,2,9,11 Reports on bacteria detected in maternal fluids during pregnancy are typically associated with adverse consequences, and whether the female reproductive tract harbours distinct microbial communities beyond the vagina has been a matter of debate. Here we systematically sample the microbiota within the female reproductive tract in 110 women of reproductive age, and examine the nature of colonisation by 16S rRNA gene amplicon sequencing and cultivation. We find distinct microbial communities in cervical canal, uterus, fallopian tubes and perito- neal fluid, differing from that of the vagina. The results reflect a microbiota continuum along the female reproductive tract, indicative of a non-sterile environment. We also identify microbial taxa and potential functions that correlate with the menstrual cycle or are over- represented in subjects with adenomyosis or infertility due to endometriosis. The study provides insight into the nature of the vagino-uterine microbiome, and suggests that sur- veying the vaginal or cervical microbiota might be useful for detection of common diseases in the upper reproductive tract. -

Modulation of Gut Microbiota by Glucosamine and Chondroitin in a Randomized, Double-Blind Pilot Trial in Humans
microorganisms Article Modulation of Gut Microbiota by Glucosamine and Chondroitin in a Randomized, Double-Blind Pilot Trial in Humans Sandi L. Navarro 1,* , Lisa Levy 1, Keith R. Curtis 1, Johanna W. Lampe 1,2 and Meredith A.J. Hullar 1 1 Division of Public Health Sciences, Fred Hutchinson Cancer Research Center, Seattle, WA 98109, USA; [email protected] (L.L.); [email protected] (K.R.C.); [email protected] (J.W.L.); [email protected] (M.A.J.H.) 2 Department of Epidemiology, University of Washington, Seattle, WA 98195, USA * Correspondence: [email protected]; Tel.: +1-206-667-6583 Received: 31 October 2019; Accepted: 22 November 2019; Published: 23 November 2019 Abstract: Glucosamine and chondroitin (G&C), typically taken for joint pain, are among the most frequently used specialty supplements by US adults. More recently, G&C have been associated with lower incidence of colorectal cancer in human observational studies and reduced severity of experimentally-induced ulcerative colitis in rodents. However, little is known about their effects on colon-related physiology. G&C are poorly absorbed and therefore metabolized by gut microbiota. G&C have been associated with changes in microbial structure, which may alter host response. We conducted a randomized, double-blind, placebo-controlled crossover trial in ten healthy adults to evaluate the effects of a common dose of G&C compared to placebo for 14 days on gut microbial community structure, measured by 16S rRNA gene sequencing. Linear mixed models were used to evaluate the effect of G&C compared to placebo on fecal microbial alpha and beta diversity, seven phyla, and 137 genera. -

Impact of Topical Antimicrobial Treatments on Skin Bacterial Communities
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations 2017 Impact Of Topical Antimicrobial Treatments On Skin Bacterial Communities Adam Sanmiguel University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Bioinformatics Commons, and the Microbiology Commons Recommended Citation Sanmiguel, Adam, "Impact Of Topical Antimicrobial Treatments On Skin Bacterial Communities" (2017). Publicly Accessible Penn Dissertations. 2567. https://repository.upenn.edu/edissertations/2567 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/2567 For more information, please contact [email protected]. Impact Of Topical Antimicrobial Treatments On Skin Bacterial Communities Abstract Skin is our primary interface to the outside world, representing a diverse habitat with a multitude of folds, invaginations, and appendages. While each of these structures is essential to host cutaneous function, they also serve as unique ecological niches that can support an array of microbial inhabitants. Together, these microorganisms constitute the skin microbiome, an assemblage of bacteria, fungi, and viruses with the potential to influence cutaneous biology. While a number of studies have described the importance of these residents to immune function and development, none to date have assessed their dynamics in response to antimicrobial stress, nor the impact of these perturbations on host cutaneous defense. Rather the majority of work in this regard has focused on a subset of microorganisms studied in isolation. Herein, we present the impact of topical antibiotics and antiseptics on skin bacterial communities, and describe their potential to shape cutaneous interactions. Using mice as a model system, we show that antibiotics can elicit a distinct shift in skin inhabitants characterized by decreases in diversity and domination by previously minor contributors.