This Electronic Thesis Or Dissertation Has Been Downloaded from the King’S Research Portal At
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genomics of Inherited Bone Marrow Failure and Myelodysplasia Michael
Genomics of inherited bone marrow failure and myelodysplasia Michael Yu Zhang A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Washington 2015 Reading Committee: Mary-Claire King, Chair Akiko Shimamura Marshall Horwitz Program Authorized to Offer Degree: Molecular and Cellular Biology 1 ©Copyright 2015 Michael Yu Zhang 2 University of Washington ABSTRACT Genomics of inherited bone marrow failure and myelodysplasia Michael Yu Zhang Chair of the Supervisory Committee: Professor Mary-Claire King Department of Medicine (Medical Genetics) and Genome Sciences Bone marrow failure and myelodysplastic syndromes (BMF/MDS) are disorders of impaired blood cell production with increased leukemia risk. BMF/MDS may be acquired or inherited, a distinction critical for treatment selection. Currently, diagnosis of these inherited syndromes is based on clinical history, family history, and laboratory studies, which directs the ordering of genetic tests on a gene-by-gene basis. However, despite extensive clinical workup and serial genetic testing, many cases remain unexplained. We sought to define the genetic etiology and pathophysiology of unclassified bone marrow failure and myelodysplastic syndromes. First, to determine the extent to which patients remained undiagnosed due to atypical or cryptic presentations of known inherited BMF/MDS, we developed a massively-parallel, next- generation DNA sequencing assay to simultaneously screen for mutations in 85 BMF/MDS genes. Querying 71 pediatric and adult patients with unclassified BMF/MDS using this assay revealed 8 (11%) patients with constitutional, pathogenic mutations in GATA2 , RUNX1 , DKC1 , or LIG4 . All eight patients lacked classic features or laboratory findings for their syndromes. -

Noncoding Rnas As Novel Pancreatic Cancer Targets
NONCODING RNAS AS NOVEL PANCREATIC CANCER TARGETS by Amy Makler A Thesis Submitted to the Faculty of The Charles E. Schmidt College of Science In Partial Fulfillment of the Requirements for the Degree of Master of Science Florida Atlantic University Boca Raton, FL August 2018 Copyright 2018 by Amy Makler ii ACKNOWLEDGEMENTS I would first like to thank Dr. Narayanan for his continuous support, constant encouragement, and his gentle, but sometimes critical, guidance throughout the past two years of my master’s education. His faith in my abilities and his belief in my future success ensured I continue down this path of research. Working in Dr. Narayanan’s lab has truly been an unforgettable experience as well as a critical step in my future endeavors. I would also like to extend my gratitude to my committee members, Dr. Binninger and Dr. Jia, for their support and suggestions regarding my thesis. Their recommendations added a fresh perspective that enriched our initial hypothesis. They have been indispensable as members of my committee, and I thank them for their contributions. My parents have been integral to my successes in life and their support throughout my education has been crucial. They taught me to push through difficulties and encouraged me to pursue my interests. Thank you, mom and dad! I would like to thank my boyfriend, Joshua Disatham, for his assistance in ensuring my writing maintained a logical progression and flow as well as his unwavering support. He was my rock when the stress grew unbearable and his encouraging words kept me pushing along. -

Somatic Mutations and Progressive Monosomy Modify SAMD9-Related Phenotypes in Humans
Downloaded from http://www.jci.org on June 26, 2017. https://doi.org/10.1172/JCI91913 RESEARCH ARTICLE The Journal of Clinical Investigation Somatic mutations and progressive monosomy modify SAMD9-related phenotypes in humans Federica Buonocore,1 Peter Kühnen,2 Jenifer P. Suntharalingham,1 Ignacio Del Valle,1 Martin Digweed,3 Harald Stachelscheid,4 Noushafarin Khajavi,2 Mohammed Didi,5 Angela F. Brady,6 Oliver Blankenstein,2 Annie M. Procter,7 Paul Dimitri,8 Jerry K.H. Wales,9 Paolo Ghirri,10 Dieter Knöbl,11 Brigitte Strahm,12 Miriam Erlacher,12,13 Marcin W. Wlodarski,12,13 Wei Chen,14 George K. Kokai,15 Glenn Anderson,16 Deborah Morrogh,17 Dale A. Moulding,18 Shane A. McKee,19 Charlotte M. Niemeyer,12,13 Annette Grüters,2 and John C. Achermann1 1Genetics and Genomic Medicine, University College London (UCL) Great Ormond Street Institute of Child Health, London, United Kingdom. 2Institute of Experimental Pediatric Endocrinology and Department of Pediatric Endocrinology, Charité, Berlin, Germany. 3Department of Human and Medical Genetics, Charité, Berlin, Germany. 4Berlin Institute of Health, Berlin, Germany, and Berlin-Brandenburg Centrum for Regenerative Therapies, Charité, Berlin, Germany. 5Department of Paediatric Endocrinology, Alder Hey Children’s NHS Foundation Trust, Liverpool, United Kingdom. 6North West Thames Regional Genetics Service, Northwick Park Hospital, Harrow, United Kingdom. 7Institute of Medical Genetics, University Hospital of Wales, Cardiff, United Kingdom. 8Academic Unit of Child Health, University of Sheffield, Sheffield, United Kingdom. 9Department of Endocrinology, Children’s Health Queensland Clinical Unit, University of Queensland, Brisbane, Australia. 10Department of Neonatology, University of Pisa, Pisa, Italy. 11Pediatric Endocrinology, Karlsruhe, Germany. -

Of Small Intestine Harboring Driver Gene Mutations: a Case Report and a Literature Review
1161 Case Report A rare multiple primary sarcomatoid carcinoma (SCA) of small intestine harboring driver gene mutations: a case report and a literature review Zhu Zhu1#, Xinyi Liu2#, Wenliang Li1, Zhengqi Wen1, Xiang Ji1, Ruize Zhou1, Xiaoyu Tuo3, Yaru Chen2, Xian Gong2, Guifeng Liu2, Yanqing Zhou2, Shifu Chen2, Lele Song2#^, Jian Huang1 1Department of Oncology, First Affiliated Hospital of Kunming Medical University, Kunming, China; 2HaploX Biotechnology, Shenzhen, China; 3Department of Pathology, First Affiliated Hospital of Kunming Medical University, Kunming, China #These authors contributed equally to this work. Correspondence to: Jian Huang. Department of Oncology, First Affiliated Hospital of Kunming Medical University, No. 295, Xichang Road, Kunming 560032, Yunnan Province, China. Email: [email protected]; Lele Song. HaploX Biotechnology, 8th floor, Auto Electric Power Building, Songpingshan Road, Nanshan District, Shenzhen 518057, Guangdong Province, China. Email: [email protected]. Abstract: Primary sarcomatoid carcinoma (SCA) is a type of rare tumor consisting of both malignant epithelial and mesenchymal components. Only 32 cases of SCA of the small bowel have been reported in the literature to date. Due to its rarity and complexity, this cancer has not been genetically studied and its diagnosis and treatment remain difficult. Here we report a 54-year-old male underwent emergency surgical resection in the small intestine due to severe obstruction and was diagnosed with multiple SCA based on postoperative pathological examination. Over 100 polypoid tumors scattered along his whole jejunum and proximal ileum. Chemotherapy (IFO+Epirubicin) was performed after surgery while the patient died two months after the surgery due to severe malnutrition. Whole-exome sequencing was performed for the tumor tissue with normal tissue as the control. -

Somatic Mutations and Progressive Monosomy Modify SAMD9-Related Phenotypes in Humans
Downloaded from http://www.jci.org on June 5, 2017. https://doi.org/10.1172/JCI91913 RESEARCH ARTICLE The Journal of Clinical Investigation Somatic mutations and progressive monosomy modify SAMD9-related phenotypes in humans Federica Buonocore,1 Peter Kühnen,2 Jenifer P. Suntharalingham,1 Ignacio Del Valle,1 Martin Digweed,3 Harald Stachelscheid,4 Noushafarin Khajavi,2 Mohammed Didi,5 Angela F. Brady,6 Oliver Blankenstein,2 Annie M. Procter,7 Paul Dimitri,8 Jerry K.H. Wales,9 Paolo Ghirri,10 Dieter Knöbl,11 Brigitte Strahm,12 Miriam Erlacher,12,13 Marcin W. Wlodarski,12,13 Wei Chen,14 George K. Kokai,15 Glenn Anderson,16 Deborah Morrogh,17 Dale A. Moulding,18 Shane A. McKee,19 Charlotte M. Niemeyer,12,13 Annette Grüters,2 and John C. Achermann1 1Genetics and Genomic Medicine, University College London (UCL) Great Ormond Street Institute of Child Health, London, United Kingdom. 2Institute of Experimental Pediatric Endocrinology and Department of Pediatric Endocrinology, Charité, Berlin, Germany. 3Department of Human and Medical Genetics, Charité, Berlin, Germany. 4Berlin Institute of Health, Berlin, Germany, and Berlin-Brandenburg Centrum for Regenerative Therapies, Charité, Berlin, Germany. 5Department of Paediatric Endocrinology, Alder Hey Children’s NHS Foundation Trust, Liverpool, United Kingdom. 6North West Thames Regional Genetics Service, Northwick Park Hospital, Harrow, United Kingdom. 7Institute of Medical Genetics, University Hospital of Wales, Cardiff, United Kingdom. 8Academic Unit of Child Health, University of Sheffield, Sheffield, United Kingdom. 9Department of Endocrinology, Children’s Health Queensland Clinical Unit, University of Queensland, Brisbane, Australia. 10Department of Neonatology, University of Pisa, Pisa, Italy. 11Pediatric Endocrinology, Karlsruhe, Germany. -

Genome-Wide Association Study for Feed Efficiency and Growth Traits in U.S. Beef Cattle Christopher M
Seabury et al. BMC Genomics (2017) 18:386 DOI 10.1186/s12864-017-3754-y RESEARCH ARTICLE Open Access Genome-wide association study for feed efficiency and growth traits in U.S. beef cattle Christopher M. Seabury1*, David L. Oldeschulte1, Mahdi Saatchi2, Jonathan E. Beever3, Jared E. Decker4,5, Yvette A. Halley1, Eric K. Bhattarai1, Maral Molaei1, Harvey C. Freetly6, Stephanie L. Hansen2, Helen Yampara-Iquise4, Kristen A. Johnson7, Monty S. Kerley4, JaeWoo Kim4, Daniel D. Loy2, Elisa Marques8, Holly L. Neibergs7, Robert D. Schnabel4,5, Daniel W. Shike3, Matthew L. Spangler9, Robert L. Weaber10, Dorian J. Garrick2,11 and Jeremy F. Taylor4* Abstract Background: Single nucleotide polymorphism (SNP) arrays for domestic cattle have catalyzed the identification of genetic markers associated with complex traits for inclusion in modern breeding and selection programs. Using actual and imputed Illumina 778K genotypes for 3887 U.S. beef cattle from 3 populations (Angus, Hereford, SimAngus), we performed genome-wide association analyses for feed efficiency and growth traits including average daily gain (ADG), dry matter intake (DMI), mid-test metabolic weight (MMWT), and residual feed intake (RFI), with marker-based heritability estimates produced for all traits and populations. Results: Moderate and/or large-effect QTL were detected for all traits in all populations, as jointly defined by the estimated proportion of variance explained (PVE) by marker effects (PVE ≥ 1.0%) and a nominal P-value threshold (P ≤ 5e-05). Lead SNPs with PVE ≥ 2.0% were considered putative evidence of large-effect QTL (n = 52), whereas those with PVE ≥ 1.0% but < 2.0% were considered putative evidence for moderate-effect QTL (n = 35). -

Bioinformatics Mining of the Dark Matter Proteome For
BIOINFORMATICS MINING OF THE DARK MATTER PROTEOME FOR CANCER TARGETS DISCOVERY by Ana Paula Delgado A Thesis Submitted to the Faculty of The Charles E. Schmidt College of Science In Partial Fulfillment of the Requirements for the Degree of Master of Science Florida Atlantic University Boca Raton, Florida May 2015 Copyright 2015 by Ana Paula Delgado ii ACKNOWLEDGEMENTS I would first like to thank Dr. Narayanan for his continuous encouragement, guidance, and support during the past two years of my graduate education. It has truly been an unforgettable experience working in his laboratory. I also want to express gratitude to my external advisor Professor Van de Ven from the University of Leuven, Belgium for his constant involvement and assistance on my project. Moreover, I would like to thank Dr. Binninger and Dr. Dawson-Scully for their advice and for agreeing to serve on my thesis committee. I also thank provost Dr. Perry for his involvement in my project. I thank Jeanine Narayanan for editorial assistance with the publications and with this dissertation. It has been a pleasure working with various undergraduate students some of whom became lab mates including Pamela Brandao, Maria Julia Chapado and Sheilin Hamid. I thank them for their expert help in the projects we were involved in. Lastly, I want to express my profound thanks to my parents and brother for their unconditional love, support and guidance over the last couple of years. They were my rock when I was in doubt and never let me give up. I would also like to thank my boyfriend Spencer Daniel and best friends for being part of an incredible support system. -

Early Mechanisms of Retinal Degeneration in the Harlequin Mouse
Western University Scholarship@Western Electronic Thesis and Dissertation Repository 7-17-2014 12:00 AM Early Mechanisms of Retinal Degeneration in the harlequin Mouse Eric Dolinar The University of Western Ontario Supervisor Dr. Kathleen Hill The University of Western Ontario Graduate Program in Biology A thesis submitted in partial fulfillment of the equirr ements for the degree in Master of Science © Eric Dolinar 2014 Follow this and additional works at: https://ir.lib.uwo.ca/etd Part of the Biology Commons, Genetics Commons, and the Molecular Genetics Commons Recommended Citation Dolinar, Eric, "Early Mechanisms of Retinal Degeneration in the harlequin Mouse" (2014). Electronic Thesis and Dissertation Repository. 2156. https://ir.lib.uwo.ca/etd/2156 This Dissertation/Thesis is brought to you for free and open access by Scholarship@Western. It has been accepted for inclusion in Electronic Thesis and Dissertation Repository by an authorized administrator of Scholarship@Western. For more information, please contact [email protected]. i Early Mechanisms of Retinal Degeneration in the harlequin Mouse (Thesis Format: Monograph) by Eric A Dolinar Graduate Program in Biology A thesis submitted in partial fulfillment of the requirements for the degree of Master of Science The School of Graduate and Postdoctoral Studies The University of Western Ontario, London, Ontario, Canada © Eric A Dolinar 2014 i ii Abstract Retinal diseases are personally debilitating and expensive, yet many early disease mechanisms leading to their onset and progression remain poorly understood. The harlequin mouse is a model of human mitochondrial dysfunction and parainflammation leading to subsequent cerebellar and retinal degeneration. Diagnosis of retinal degeneration can be tracked in vivo and is associated with AIF dysfunction. -

Supplementary Data
Gene expression profile of the A549 human non-small cell lung carcinoma cell line following treatment with the seeds of Descurainia sophia, a potential anticancer drug Bu-Yeo Kim, Jun Lee, Sung Joon Park, Ok-Sun Bang, and No Soo Kim Supplementary Data Figure Legends Supplementary Figure 1. Electropherogram of total RNA samples used for gene expression profiles. The A549 cells were treated with increasing concentrations of EEDS (0-20 mg/mL). After 24 h drug treatment, total RNAs were extracted from the cells, and their integrities were determined by electropherogram. RIN values range from 10 (intact) to 1(totally degraded). Supplementary Figure 2. Pathways enriched (FDR<0.01) in the Up- and Down-patterns. The genes that are shown in Table 5 and Figure 4, and involved in each pathway are highlighted in green Supplementary Figure 3. The level of similarity is represented in red with a scale bar. The color in the box of a diagonal line or “Activity” on the right panel represents the activity of the pathway. The positions and names of signaling-related pathways are colored red and the metabolism-related pathways are colored blue. Supplementary Table 1. Quality control of total RNA samples used for gene expression analysis. * EEDS (mg/mL) OD260/280 OD260/230 Ratio (28s/18s) RIN Result 0 2.04 2.22 2.3 9.8 Pass 1.25 2.03 2.28 2.2 10.0 Pass 5 2.03 2.28 2.1 10.0 Pass 20 2.04 2.23 2.0 10.0 Pass *RIN values, 10 (intact) to 1 (completely degraded). -

Deciphering Common and Rare Genetic E Ects on Reading Ability
Deciphering common and rare genetic eects on reading ability © 2016, Amaia Carrion´ Castillo ISBN: 978-90-76203-78-2 Printed and bound by Ipskamp Drukkers Deciphering common and rare genetic eects on reading ability Proefschri ter verkrijging van de graad van doctor aan de Radboud Universiteit Nijmegen op gezag van de rector magnicus prof. dr. J.H.J.M. van Krieken, volgens besluit van het college van decanen in het openbaar te verdedigen op maandag 7 november 2016 om 12.30 uur precies door Amaia Carrion´ Castillo geboren op 22 november 1988 te Donostia (Spanje) Promotor Prof. dr. Simon E. Fisher Copromotor Dr. Clyde Francks (MPI) Manuscriptcommissie Prof. dr. Anneke I. den Hollander Prof. dr. Juha Kere (Karolinska Institutet, Zweden) Prof. dr. Gerd Schulte-Korne¨ (Ludwig-Maximilians-University Munich, Duitsland) Deciphering common and rare genetic eects on reading ability Doctoral esis to obtain the degree of doctor from Radboud University Nijmegen on the authority of the Rector Magnicus prof. dr. J.H.J.M. van Krieken, according to the decision of the Council of Deans to be defended in public on Monday, November 7, 2016 at 12.30 hours by Amaia Carrion´ Castillo Born on 22 November 1988 in Donostia (Spain) Supervisor Prof. dr. Simon E. Fisher Co-supervisor Dr. Clyde Francks (MPI) Doctoral esis Committee Prof. dr. Anneke I. den Hollander Prof. dr. Juha Kere (Karolinska Institutet, Sweden) Prof. dr. Gerd Schulte-Korne¨ (Ludwig-Maximilians-University Munich, Germany) Etxekoei CONTENTS 1 General introduction 11 1.1 e learning-to-read brain ........................ 11 1.2 Variation in reading ability........................ 13 1.3 Complex and multifactorial aetiology................. -
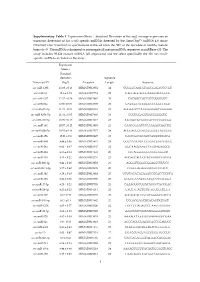
Supplementary Table 1. Expression
Supplementary Table 1. Expression (Mean Standard Deviation of the log2 average expression or transcript detection) of Sus scrofa specific miRNAs detected by the GeneChip™ miRNA 4.0 Array (ThermoFisher Scientific) in spermatozoa retrieved from the SRF of the ejaculate of healthy mature boars (n=3). The miRNA is designed to interrogate all mature miRNA sequences in miRBase v20. The array includes 30.424 mature miRNA (all organisms) and we select specifically the 326 Sus scrofa- specific miRNAs included in the array. Expression Mean ± Standard Deviation Sequence Transcript ID (log2) Accession Length Sequence ssc-miR-1285 13.98 ± 0.13 MIMAT0013954 24 CUGGGCAACAUAGCGAGACCCCGU ssc-miR-16 12.6 ± 0.74 MIMAT0007754 22 UAGCAGCACGUAAAUAUUGGCG ssc-miR-4332 12.32 ± 0.29 MIMAT0017962 20 CACGGCCGCCGCCGGGCGCC ssc-miR-92a 12.06 ± 0.09 MIMAT0013908 22 UAUUGCACUUGUCCCGGCCUGU ssc-miR-671-5p 11.73 ± 0.54 MIMAT0025381 24 AGGAAGCCCUGGAGGGGCUGGAGG ssc-miR-4334-5p 11.31 ± 0.05 MIMAT0017966 19 CCCUGGAGUGACGGGGGUG ssc-miR-425-5p 10.99 ± 0.15 MIMAT0013917 23 AAUGACACGAUCACUCCCGUUGA ssc-miR-191 10.57 ± 0.22 MIMAT0013876 23 CAACGGAAUCCCAAAAGCAGCUG ssc-miR-92b-5p 10.53 ± 0.18 MIMAT0017377 24 AGGGACGGGACGCGGUGCAGUGUU ssc-miR-15b 10.01 ± 0.9 MIMAT0002125 22 UAGCAGCACAUCAUGGUUUACA ssc-miR-30d 9.89 ± 0.36 MIMAT0013871 24 UGUAAACAUCCCCGACUGGAAGCU ssc-miR-26a 9.62 ± 0.47 MIMAT0002135 22 UUCAAGUAAUCCAGGAUAGGCU ssc-miR-484 9.55 ± 0.14 MIMAT0017974 20 CCCAGGGGGCGACCCAGGCU ssc-miR-103 9.53 ± 0.22 MIMAT0002154 23 AGCAGCAUUGUACAGGGCUAUGA ssc-miR-296-3p 9.41 ± 0.26 MIMAT0022958 -

Position Effect on FGF13 Associated with X-Linked Congenital Generalized Hypertrichosis
Position effect on FGF13 associated with X-linked congenital generalized hypertrichosis Gina M. DeStefanoa, Katherine A. Fantauzzoa,b, Lynn Petukhovab,c, Mazen Kurbanb, Marija Tadin-Strappsa, Brynn Levyd, Dorothy Warburtona,e, Elizabeth T. Cirullif, Yujun Hanf, Xiaoyun Sung, Yufeng Sheng, Maryam Shirazid, Vaidehi Jobanputrad, Rodrigo Cepeda-Valdesh, Julio Cesar Salas-Alanish,i, and Angela M. Christianoa,b,1 Departments of aGenetics and Development, bDermatology, cEpidemiology, and dPathology and Cell Biology, Columbia University, New York, NY; eDepartment of Pediatrics, Columbia University Medical Center, New York, NY 10032; fCenter for Human Genome Variation, Duke University School of Medicine, Durham, NC 27708; gDepartment of Biomedical Informatics, Columbia Initiative in Systems Biology, Columbia University, New York, NY 10032; hDystrophic Epidermolysis Bullosa Research Association (DebRA), Nuevo Leon 67150, Mexico; and iBasic Science, Universidad de Monterrey, Nueva Leon 63238, Mexico Edited by Elaine Fuchs, The Rockefeller University, New York, NY, and approved March 12, 2013 (received for review October 17, 2012) X-linked congenital generalized hypertrichosis (Online Mendelian providing evidence to support a position effect as the un- Inheritance in Man 307150) is an extremely rare condition of hair derlying genetic basis of X-linked hypertrichosis. overgrowth on different body sites. We previously reported link- age in a large Mexican family with X-linked congenital generalized Results and Discussion hypertrichosis cosegregating with deafness and with dental and We ascertained a large kindred from Mexico with X-linked CGH (Online Mendelian Inheritance in Man 307150) cosegregating palate anomalies to Xq24-27. Using SNP oligonucleotide microar- – ray analysis and whole-genome sequencing, we identified a 389- with deafness and with dental and palate anomalies (Fig.