Atomic Weights of the Elements: Review 2000
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Exploring Density
Exploring Density Students investigate the densities of different liquids and solids and understand how density may help identify a substance. Suggested Grade Range: 6-8 Approximate Time: 1 hour Relevant National Content Standards: Next Generation Science Standards Science and Engineering Practices: Developing and using Models Modeling in 6–8 builds on K–5 experiences and progresses to developing, using, and revising models to describe, test, and predict more abstract phenomena and design systems. • Develop and use a model to describe phenomena. Science and Engineering Practices: Analyzing and Interpreting Data Analyzing data in 6-8 builds on K-5 and progresses to extending quantitative analysis to investigations, distinguishing between correlation and causation, and basic statistical techniques of data and error analysis. • Analyze and interpret data to determine similarities and differences in findings. Disciplinary Core Ideas: PS1.A Structure and Properties of Matter Each pure substance has characteristic physical and chemical properties (for any bulk quantity under given conditions) that can be used to identify it. Common Core State Standard: 7NS 2. Apply and extend previous understanding of multiplication and division and of fractions to multiply and divide rational numbers. 3. Solve real-world and mathematical problems involving the four operations with rational numbers. Common Core State Standard: 7EE Solve real-life and mathematical problems using numerical and algebraic expressions and equations. 4. Use variables to represent -

Glossary of Terms
GLOSSARY OF TERMS For the purpose of this Handbook, the following definitions and abbreviations shall apply. Although all of the definitions and abbreviations listed below may have not been used in this Handbook, the additional terminology is provided to assist the user of Handbook in understanding technical terminology associated with Drainage Improvement Projects and the associated regulations. Program-specific terms have been defined separately for each program and are contained in pertinent sub-sections of Section 2 of this handbook. ACRONYMS ASTM American Society for Testing Materials CBBEL Christopher B. Burke Engineering, Ltd. COE United States Army Corps of Engineers EPA Environmental Protection Agency IDEM Indiana Department of Environmental Management IDNR Indiana Department of Natural Resources NRCS USDA-Natural Resources Conservation Service SWCD Soil and Water Conservation District USDA United States Department of Agriculture USFWS United States Fish and Wildlife Service DEFINITIONS AASHTO Classification. The official classification of soil materials and soil aggregate mixtures for highway construction used by the American Association of State Highway and Transportation Officials. Abutment. The sloping sides of a valley that supports the ends of a dam. Acre-Foot. The volume of water that will cover 1 acre to a depth of 1 ft. Aggregate. (1) The sand and gravel portion of concrete (65 to 75% by volume), the rest being cement and water. Fine aggregate contains particles ranging from 1/4 in. down to that retained on a 200-mesh screen. Coarse aggregate ranges from 1/4 in. up to l½ in. (2) That which is installed for the purpose of changing drainage characteristics. -

Carbon Dioxide
Safetygram 18 Carbon dioxide Carbon dioxide is nonflammable, colorless, and odorless in the gaseous and liquid states. Carbon dioxide is a minor but important constituent of the atmosphere, averaging about 0.036% or 360 ppm by volume. It is also a normal end-prod- uct of human and animal metabolism. Dry carbon dioxide is a relatively inert gas. In the event moisture is present in high concentrations, carbonic acid may be formed and materials resistant to this acid should be used. High flow rates or rapid depressurization of a system can cause temperatures approaching the sublimation point (–109.3°F [–78.5°C]) to be attained within the system. Carbon dioxide will convert directly from a liquid to a solid if the liquid is depressurized below 76 psia (61 psig). The use of ma- terials which become brittle at low temperatures should be avoided in applications where temperatures less than –20°F (–29°C) are expected. Vessels and piping used in carbon dioxide service should be designed to the American Society of Mechanical Engineers (ASME) or Department of Transportation (DOT) codes for the pressures and temperatures involved. Physical properties are listed in Table 1. Carbon dioxide in the gaseous state is colorless and odorless and not easily detectable. Gaseous carbon dioxide is 1.5 times denser than air and therefore is found in greater concentrations at low levels. Ventilation systems should be designed to exhaust from the lowest levels and allow make-up air to enter at a higher level. Manufacture Carbon dioxide is produced as a crude by-product of a number of manufactur- ing processes. -

Guidelines for the Use of Atomic Weights 5 10 11 12 DOI: ..., Received ...; Accepted
IUPAC Guidelines for the us e of atomic weights For Peer Review Only Journal: Pure and Applied Chemistry Manuscript ID PAC-REC-16-04-01 Manuscript Type: Recommendation Date Submitted by the Author: 01-Apr-2016 Complete List of Authors: van der Veen, Adriaan; VSL Meija, Juris Possolo, Antonio; National Institute of Standards and Technology Hibbert, David; University of New South Wales, School of Chemistry atomic weights, atomic-weight intervals, molecular weight, standard Keywords: atomic weight, measurement uncertainty, uncertainty propagation Author-Supplied Keywords: P.O. 13757, Research Triangle Park, NC (919) 485-8700 Page 1 of 13 IUPAC Pure Appl. Chem. 2016; aop 1 2 3 4 Sponsoring body: IUPAC Inorganic Chemistry Division Committee: see more details on page XXX. 5 IUPAC Recommendation 6 7 Adriaan M. H. van der Veen*, Juris Meija, Antonio Possolo, and D. Brynn Hibbert 8 9 Guidelines for the use of atomic weights 5 10 11 12 DOI: ..., Received ...; accepted ... 13 14 Abstract: Standard atomicFor weights Peer are widely used Review in science, yet the uncertainties Only associated with these 15 values are not well-understood. This recommendation provides guidance on the use of standard atomic 16 weights and their uncertainties. Furthermore, methods are provided for calculating standard uncertainties 17 of molecular weights of substances. Methods are also outlined to compute material-specific atomic weights 10 18 whose associated uncertainty may be smaller than the uncertainty associated with the standard atomic 19 weights. 20 21 Keywords: atomic weights; atomic-weight intervals; molecular weight; standard atomic weight; uncertainty; 22 uncertainty propagation 23 24 25 1 Introduction 15 26 27 Atomic weights provide a practical link the SI base units kilogram and mole. -

The Water Molecule
Seawater Chemistry: Key Ideas Water is a polar molecule with the remarkable ability to dissolve more substances than any other natural solvent. Salinity is the measure of dissolved inorganic solids in water. The most abundant ions dissolved in seawater are chloride, sodium, sulfate, and magnesium. The ocean is in steady state (approx. equilibrium). Water density is greatly affected by temperature and salinity Light and sound travel differently in water than they do in air. Oxygen and carbon dioxide are the most important dissolved gases. 1 The Water Molecule Water is a polar molecule with a positive and a negative side. 2 1 Water Molecule Asymmetry of a water molecule and distribution of electrons result in a dipole structure with the oxygen end of the molecule negatively charged and the hydrogen end of the molecule positively charged. 3 The Water Molecule Dipole structure of water molecule produces an electrostatic bond (hydrogen bond) between water molecules. Hydrogen bonds form when the positive end of one water molecule bonds to the negative end of another water molecule. 4 2 Figure 4.1 5 The Dissolving Power of Water As solid sodium chloride dissolves, the positive and negative ions are attracted to the positive and negative ends of the polar water molecules. 6 3 Formation of Hydrated Ions Water dissolves salts by surrounding the atoms in the salt crystal and neutralizing the ionic bond holding the atoms together. 7 Important Property of Water: Heat Capacity Amount of heat to raise T of 1 g by 1oC Water has high heat capacity - 1 calorie Rocks and minerals have low HC ~ 0.2 cal. -

Measuring Density
Measuring Density Background All matter has mass and volume. Mass is a measure of the amount of matter an object has. Its measure is usually given in grams (g) or kilograms (kg). Volume is the amount of space an object occupies. There are numerous units for volume including liters (l), meters cubed (m3), and gallons (gal). Mass and volume are physical properties of matter and may vary with different objects. For example, it is possible for two pieces of metal to be made out of the same material yet for one piece to be bigger than the other. If the first piece of metal is twice as large as the second, then you would expect that this piece is also twice as heavy (or have twice the mass) as the first. If both pieces of metal are made of the same material the ratio of the mass and volume will be the same. We define density (ρ) as the ratio of the mass of an object to the volume it occupies. The equation is given by: M ρ = (1.1) V here the symbol M stands for the mass of the object, and V the volume. Density has the units of mass divided by volume such as grams per centimeters cube (g/cm3) or kilograms per liter (kg/l). Sample Problem #1 A block of wood has a mass of 8 g and occupies a volume of 10 cm3. What is its density? Solution 8g The density will be = 0.8g / cm 3 . 10cm 3 This means that every centimeter cube of this wood will have a mass of 0.8 grams. -

Density Objective Learn How to Calculate Density
Density Objective Learn how to calculate density Density is a way of comparing the mass of an object to its size (volume). If an object feels heavy for its size, it has a high density. A dense object has more material (atoms) packed into a smaller area. Density cannot be measured directly, it must be calculated. The formula used to calculate density is density = mass/volume or D = m/v. The typical label for density is g/mL or g/cm3. Density is a useful tool for scientist. It is often used to help identify a pure substance. A pure substance will have one unique density. For example, each element on the periodic table has a density from any other element. If you had an unknown gas, you could calculate the density of it to identify it. The same is true for minerals- though several minerals may have the same density so other properties would also need to be used to identify the mineral. If you have ever noticed oil floating on water, that is an example of density. Objects that are less dense than water, it will float. Waters density is 1.0g/mL. 1. In general terms, describe what density is. 2. How could density be used to identify an unknown substance? 3. Density must be calculated using a math formula. What is the formula for density? 4. What is a typical label for density? *Obviously both water and ice are the same substance- so should have the same density. But does it? Then why does ice float? Suggest an explanation. -

CALCULATING DENSITY Density of Common Metals Copper 8.96 G/Cm
NAME __________________________________________ PER _______________ CALCULATING DENSITY Density of Common Metals Copper 8.96 g/cm3 Gold 19.32 g/cm3 Iron 7.87 g/cm3 Lead 11.36 /cm3 1. A substance has a mass of 1370.3 grams (g) and a volume of 71 cubic centimeters (cm3). Using the table above, what is this substance? A Iron B Lead C Gold D Copper 2. A substance has a mass of 375 grams (g) and a volume of 47.65 cm3. Using the table above, what is this substance? A Iron B Lead C Gold D Copper For questions 3 – 9, calculate the density and fill in the table. 10. What has the greater density, a cube of water measuring 1cmX1cmX1cm and having a mass of 1g, or a block of plastic measuring 2cmX3cmX1cm with a mass of 4g? A Cube of water B Block of plastic 11. A rock has a volume 57 cm3. Its mass is 14. Sam has to find the density of an 555.75 g. What is the density of this rock? irregularly shaped solid. The mass of the solid is 300.3 g. She uses water displacement to find the volume. The volume of the water alone is 100 mL. The volume of the water with the solid in it is 142 mL. What is the density of the solid? 12. Joe has to find the density of a rectangular box. He measures the width to be 6 cm, the length to be 7 cm, and the height to be 2 cm. He put the box on a balance and the mass is 15. -
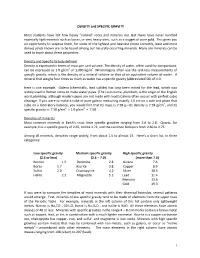
DENSITY and SPECIFIC GRAVITY Most Students Have Felt How Heavy
DENSITY and SPECIFIC GRAVITY Most students have felt how heavy “normal” rocks and minerals are. But many have never handled especially light minerals such as borax, or very heavy ones, such as a nugget of pure gold. This gives you an opportunity to surprise them, for some of the lightest and heaviest (more correctly, least and most dense) solids known are to be found among our naturally occurring minerals. Many ore minerals can be used to teach about these properties. Density and Specific Gravity defined Density is expressed in terms of mass per unit volume. The density of water, often used for comparisons, can be expressed as 1.0 g/cm3 or 1,000 kg/m3. Mineralogists often use the unit-less measurements of specific gravity, which is the density of a mineral relative to that of an equivalent volume of water. A mineral that weighs four times as much as water has a specific gravity (abbreviated SG) of 4.0. Here is one example. Galena (chemically, lead sulfide) has long been mined for the lead, which was widely used in Roman times to make water pipes. (The Latin name, plumbum, is the origin of the English word plumbing, although modern pipes are not made with lead.) Galena often occurs with perfect cubic cleavage. If you were to make a cube of pure galena measuring exactly 1.0 cm on a side and place that cube on a laboratory balance, you would find that its mass is 7.58 g—its density is 7.58 g/cm3, and its specific gravity is: 7.58 g/cm3 ÷ 1.0 g/cm3 = 7.58 Densities of minerals Most common minerals in Earth’s crust have specific gravities ranging from 2.6 to 2.8. -

International Alloy Designations and Chemical Composition Limits for Wrought Aluminum and Wrought Aluminum Alloys
International Alloy Designations and Chemical Composition Limits for Wrought Aluminum and Wrought Aluminum Alloys 1525 Wilson Boulevard, Arlington, VA 22209 www.aluminum.org With Support for On-line Access From: Aluminum Extruders Council Australian Aluminium Council Ltd. European Aluminium Association Japan Aluminium Association Alro S.A, R omania Revised: January 2015 Supersedes: February 2009 © Copyright 2015, The Aluminum Association, Inc. Unauthorized reproduction and sale by photocopy or any other method is illegal . Use of the Information The Aluminum Association has used its best efforts in compiling the information contained in this publication. Although the Association believes that its compilation procedures are reliable, it does not warrant, either expressly or impliedly, the accuracy or completeness of this information. The Aluminum Association assumes no responsibility or liability for the use of the information herein. All Aluminum Association published standards, data, specifications and other material are reviewed at least every five years and revised, reaffirmed or withdrawn. Users are advised to contact The Aluminum Association to ascertain whether the information in this publication has been superseded in the interim between publication and proposed use. CONTENTS Page FOREWORD ........................................................................................................... i SIGNATORIES TO THE DECLARATION OF ACCORD ..................................... ii-iii REGISTERED DESIGNATIONS AND CHEMICAL COMPOSITION -

Eureka! Density! by Cindy Grigg
Eureka! Density! By Cindy Grigg 1 What is density? Density is simply the amount of "stuff" in a given space. Scientists measure density by dividing the mass of something by its volume (d = m/v). This is a story about how the concept of density was first "discovered." 2 It is the story of a Greek mathematician named Archimedes who lived around 250 B.C. The King of Syracuse, where Archimedes lived, thought that he was being cheated by the metal craftsman who made his golden crown. The King called Archimedes to him and gave him the task of finding out whether the craftsman had replaced some of the gold in the King's crown with silver. Silver was worth less money than gold, and it also was an insult to the King to be wearing a crown that was not pure gold. 3 The King gave Archimedes some rules. Archimedes could not damage the crown in any way. He could not melt down the crown to see if it was made of other metals. He could not scratch the crown to see if there was silver underneath the golden outside. Archimedes thought about the problem while taking a bath. As he entered the bathing pool, he noticed that water spilled over the sides of the pool. He realized that the amount of water that spilled was equal in volume to the space that his body occupied. This fact suddenly provided him with a method for finding out if the King's crown was made of pure gold. 4 Archimedes knew that silver is not as "heavy" as gold. -

Handbook for the Application of the Caesium-137 Technique
Use of Caesium - 3 7 as a Tracer of XA04N1922 Erosion and Sedimentation: Handbook for the Application of the Caesium-137 Technique fNIS-XA-N-- 79 UK Overseas Development Administration Research Scheme R4579 .4T. te, October 1993 D.E. Walling C,- TA. uIlIc Department o'Geqqi-apky, nhei-slty of Exctc;-. Use of Caesium-137 as a Tracer of Erosion and Sedimentation: Handbook for the Application of the Caesium-137 Technique. UK Overseas Development Administration Research Scheme R4579 D.E.Walling & T.A.Ouine October 1993 Department of Geography University of Exeter Contents Part I Methodology Chapter Soil Erosion - The Problem and its Assessment 1.1 The Impacts of Soil Erosion 1.1.1 On-site impact on agricultural productivitV. 1.1.2 Off-siteimpactsoterodedsediment. 1.2 The Data Requirement 1.2.1 Data required to assess the impact on productiv 1.2.2 Data required to assess the off-site impact 1.2.3 Data required to target conservation resources 1.3 Current Methods of Erosion Assessment and the Resultant Data 1.3.1 Long-term monitoring of experimental plots 1.3.2 Field survey of erosion features 1.3.3 Erosion modelling 1.4 Meeting the Data Requirements 1.5 The Potential of the Caesium- 137 Technique 1.6 References Chapter 2 The Caesium-137 Technique 2.1 The Basis of the Caesium-137 Technique 2.2 The Source of Caesium-137 in the Environment 2.3 Deposition of Caesium-137 2.3.1 Bomb-derived caesium-137 2.3.2 ChernobVI-derived caesium-137 2.4 Caesium-137 adsorption by mineral sediment 2.4.1 Experimental studies 2.4.2 Field studies 2.5 Sediment-associated redistribution of caesium-1 37 2.5.1 Field evidence 2.6 Use of Caesium-137 Measurements in Erosion Assessment 2.7 Establishment of a Reference Inventory 2.8 Measurement of the Spatial Distribution of Caesiurn- 1 37 2.9 Identification of Caesium-137 Redistribution 2.10 Development of 'Calibration' Procedures 2.11 Estimation of Erosion and Aggradation Rates 2.12 Summary 2.13 References Chapter 3 Sample Collection, Preparation and Analysis 3.1 Sample Collection 3.