Preparation and Lewis Acid Properties of Diboron Tetrafluoride and Related Compounds
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
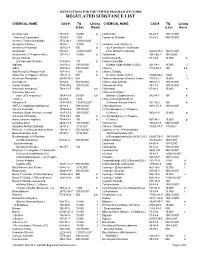
Regulated Substance List
INSTRUCTIONS FOR THE UNIFIED PROGRAM (UP) FORM REGULATED SUBSTANCE LIST CHEMICAL NAME CAS # TQ Listing CHEMICAL NAME CAS # TQ Listing (Lbs) Basis (Lbs) Basis Acetaldehyde 75-07-0 10,000 g Cantharidin 56-25-7 100/10,0001 * Acetone Cyanohydrin 75-86-5 1,000 Carbachol Chloride 51-83-2 500/10,0001 Acetone Thiosemicarbazide 1752-30-3 1,000/10,0001 Acetylene (Ethyne) 74-86-2 10,000 f Carbamic Acid, Methyl-,o- Acrolein (2-Propenal) 107-02-8 500 b (((2,4-Dimethyl-1,3-Dithiolan- Acrylamide 79-06-1 1,000/10,0001 2-YL) Methylene)Amino)- 26419-73-8 100/10,0001 Acrylonitrile (2- Propenenitrile) 107-13-1 10,000 b Carbofuran 1563-66-2 10/10,0001 Acrylyl Chloride Carbon Disulfide 75-15-0 10,000 b (2-Propenoyl Chloride) 814-68-6 100 b Carbon Oxysulfide Aldicarb 116-06-3 100/10,0001 (Carbon Oxide Sulfide (COS)) 463-58-1 10,000 f Aldrin 309-00-2 500/10,0001 Chlorine 7782-50-5 100 a,b Allyl Alcohol (2-Propen-1-ol) 107-18-6 1,000 b Chlorine Dioxide Allylamine (2-Propen-1-Amine) 107-11-9 500 b (Chlorine Oxide (ClO2)) 10049-04-4 1,000 c Aluminum Phosphide 20859-73-8 500 Chlorine Monoxide (Chlorine Oxide) 7791-21-1 10,000 f Aminopterin 54-62-6 500/10,0001 Chlormequat Chloride 999-81-5 100/10,0001 Amiton Oxalate 3734-97-2 100/10,0001 Chloroacetic Acid 79-11-8 100/10,0001 Ammonia, Anhydrous 2 7664-41-7 500 a,b Chloroform 67-66-3 10,000 b Ammonia, Aqueous Chloromethyl Ether (conc 20% or greater) 7664-41-7 20,000 a,b (Methane,Oxybis(chloro-) 542-88-1 100 b * Aniline 62-53-3 1,000 Chloromethyl Methyl Ether Antimycin A 1397-94-0 1,000/10,0001 (Chloromethoxymethane) -

Thermodynamic Hydricity of Small Borane Clusters and Polyhedral Closo-Boranes
molecules Article Thermodynamic Hydricity of Small Borane Clusters y and Polyhedral closo-Boranes Igor E. Golub 1,* , Oleg A. Filippov 1 , Vasilisa A. Kulikova 1,2, Natalia V. Belkova 1 , Lina M. Epstein 1 and Elena S. Shubina 1,* 1 A. N. Nesmeyanov Institute of Organoelement Compounds and Russian Academy of Sciences (INEOS RAS), 28 Vavilova St, 119991 Moscow, Russia; [email protected] (O.A.F.); [email protected] (V.A.K.); [email protected] (N.V.B.); [email protected] (L.M.E.) 2 Faculty of Chemistry, M.V. Lomonosov Moscow State University, 1/3 Leninskiye Gory, 119991 Moscow, Russia * Correspondence: [email protected] (I.E.G.); [email protected] (E.S.S.) Dedicated to Professor Bohumil Štibr (1940-2020), who unfortunately passed away before he could reach the y age of 80, in the recognition of his outstanding contributions to boron chemistry. Academic Editors: Igor B. Sivaev, Narayan S. Hosmane and Bohumír Gr˝uner Received: 6 June 2020; Accepted: 23 June 2020; Published: 25 June 2020 MeCN Abstract: Thermodynamic hydricity (HDA ) determined as Gibbs free energy (DG◦[H]−) of the H− detachment reaction in acetonitrile (MeCN) was assessed for 144 small borane clusters (up 2 to 5 boron atoms), polyhedral closo-boranes dianions [BnHn] −, and their lithium salts Li2[BnHn] (n = 5–17) by DFT method [M06/6-311++G(d,p)] taking into account non-specific solvent effect (SMD MeCN model). Thermodynamic hydricity values of diborane B2H6 (HDA = 82.1 kcal/mol) and its 2 MeCN dianion [B2H6] − (HDA = 40.9 kcal/mol for Li2[B2H6]) can be selected as border points for the range of borane clusters’ reactivity. -

Borontrifluoride Etherate Safety Data Sheet 6160301 According to Federal Register / Vol
Borontrifluoride etherate Safety Data Sheet 6160301 according to Federal Register / Vol. 77, No. 58 / Monday, March 26, 2012 / Rules and Regulations Date of issue: 02/09/2016 Version: 1.0 SECTION 1: Identification 1.1. Identification Product form : Substance Substance name : Borontrifluoride etherate CAS No : 109-63-7 Product code : 6160-3-01 Formula : C4H10BF3O Synonyms : Boron trifluoride diethyl etherate Other means of identification : MFCD00013194 1.2. Relevant identified uses of the substance or mixture and uses advised against Use of the substance/mixture : Laboratory chemicals Manufacture of substances Scientific research and development 1.3. Details of the supplier of the safety data sheet SynQuest Laboratories, Inc. P.O. Box 309 Alachua, FL 32615 - United States of America T (386) 462-0788 - F (386) 462-7097 [email protected] - www.synquestlabs.com 1.4. Emergency telephone number Emergency number : (844) 523-4086 (3E Company - Account 10069) SECTION 2: Hazard(s) identification 2.1. Classification of the substance or mixture Classification (GHS-US) Flam. Liq. 3 H226 - Flammable liquid and vapour Acute Tox. 4 (Oral) H302 - Harmful if swallowed Acute Tox. 1 (Inhalation) H330 - Fatal if inhaled Skin Corr. 1A H314 - Causes severe skin burns and eye damage Eye Dam. 1 H318 - Causes serious eye damage STOT SE 3 H335 - May cause respiratory irritation STOT RE 1 H372 - Causes damage to organs through prolonged or repeated exposure Full text of H-phrases: see section 16 2.2. Label elements GHS-US labeling Hazard pictograms (GHS-US) : GHS02 GHS05 GHS06 GHS07 GHS08 Signal word (GHS-US) : Danger Hazard statements (GHS-US) : H226 - Flammable liquid and vapor H302 - Harmful if swallowed H314 - Causes severe skin burns and eye damage H330 - Fatal if inhaled H335 - May cause respiratory irritation H372 - Causes damage to organs through prolonged or repeated exposure Precautionary statements (GHS-US) : P210 - Keep away from heat/sparks/open flames/hot surfaces. -

PATENT OFFICE PROPONCACD SYNTHESS Donald J
Patented Aug. 29, 1939 2,170,825 UNITED STATES 2,170,825PATENT OFFICE PROPONCACD SYNTHESS Donald J. Loder, Wilmington, Del, assignor to E. I. du Pont de Nenours is mington, Del, a corporation ofCompany, Delaware Wi No Drawing. Application January 27, 1938, Serial No. 18,226 8. Claims. (C. 260-532) This invention relates to an improved, process for the preparation of propionic acid and its ethyl chloride, and the like, may likewise be used, esters and particularly to the preparation of pro although due to its availability, low cost, and pionic acid by the interaction of ethanol and car adaptability I prefer to employ ethanol as the bon monoxide in the presence of boron fluoride major raw material. as the catalyst. An aqueous solution of borontrifluoride is pre An object of the invention is to provide a ferred as the catalyst, which may be made in var 5 process for the preparation of propionic acid in , ious ways such, for example, as by the solution exceedingly high yields from ethanol and carbon of boron trifluoride in water; by the interaction O monoxide. A further object of the invention is of anhydrous hydrogen fluoride with boric oxide to provide a process for preparing propionic acid or boric anhydride; or, if desired, by the intro O and its ethyl ester from ethanol and carbon mon duction of boron trifluoride as a gas directly into oxide in the presence of such proportions of water the mixture of water, ethanol, ether and/or ester and boron trifluoride that substantially 100% prior to or during the reaction. -

Refrigerant Safety Refrigerant History
Refrigerant Safety The risks associated with the use of refrigerants in refrigeration and airconditioning equipment can include toxicity, flammability, asphyxiation, and physical hazards. Although refrigerants can pose one or more of these risks, system design, engineering controls, and other techniques mitigate this risk for the use of refrigerant in various types of equipment. Refrigerant History Nearly all of the historically used refrigerants were flammable, toxic, or both. Some were also highly reactive, resulting in accidents (e. g., leak, explosion) due to equipment failure, poor maintenance, or human error. The task of finding a nonflammable refrigerant with good stability was given to Thomas Midgley in 1926. With his associates Albert Leon Henne and Robert Reed McNary, Dr. Midgley observed that the refrigerants then in use comprised relatively few chemical elements, many of which were clustered in an intersecting row and column of the periodic table of elements. The element at the intersection was fluorine, known to be toxic by itself. Midgley and his collaborators felt, however, that compounds containing fluorine could be both nontoxic and nonflammable. The attention of Midgley and his associates was drawn to organic fluorides by an error in the literature that showed the boiling point for tetrafluoromethane (carbon tetrafluoride) to be high compared to those for other fluorinated compounds. The correct boiling temperature later was found to be much lower. Nevertheless, the incorrect value was in the range sought and led to evaluation of organic fluorides as candidates. The shorthand convention, later introduced to simplify identification of the organic fluorides for a systematic search, is used today as the numbering system for refrigerants. -

Assessment of Portable HAZMAT Sensors for First Responders
The author(s) shown below used Federal funds provided by the U.S. Department of Justice and prepared the following final report: Document Title: Assessment of Portable HAZMAT Sensors for First Responders Author(s): Chad Huffman, Ph.D., Lars Ericson, Ph.D. Document No.: 246708 Date Received: May 2014 Award Number: 2010-IJ-CX-K024 This report has not been published by the U.S. Department of Justice. To provide better customer service, NCJRS has made this Federally- funded grant report available electronically. Opinions or points of view expressed are those of the author(s) and do not necessarily reflect the official position or policies of the U.S. Department of Justice. Assessment of Portable HAZMAT Sensors for First Responders DOJ Office of Justice Programs National Institute of Justice Sensor, Surveillance, and Biometric Technologies (SSBT) Center of Excellence (CoE) March 1, 2012 Submitted by ManTech Advanced Systems International 1000 Technology Drive, Suite 3310 Fairmont, West Virginia 26554 Telephone: (304) 368-4120 Fax: (304) 366-8096 Dr. Chad Huffman, Senior Scientist Dr. Lars Ericson, Director UNCLASSIFIED This project was supported by Award No. 2010-IJ-CX-K024, awarded by the National Institute of Justice, Office of Justice Programs, U.S. Department of Justice. The opinions, findings, and conclusions or recommendations expressed in this publication are those of the author(s) and do not necessarily reflect those of the Department of Justice. This document is a research report submitted to the U.S. Department of Justice. This report has not been published by the Department. Opinions or points of view expressed are those of the author(s) and do not necessarily reflect the official position or policies of the U.S. -

United States Patent Office Patented May 9, 1961 1
2,983,583 United States Patent Office Patented May 9, 1961 1. 2 of the tube and drained into the hot Zone. Most of the 2,983,583 silicon tetrachloride was recovered unchanged. 0.43 millimole of BCls were obtained. Based on the SiCl4 METHOD OF PREPARNG BORON TRICHLORDE consumed, the yield of boron trichloride was about 70% FROM BORIC OXDE AND SILICON TETRA 5 based on Equation 2. CHLORDE Example III-In another experiment, conducted in William H. Schechter, Bradford Woods, Pa., assignor to a manner similar to those above, 10.0 millimoles of S2Cl2 Callery Chemical Company, Pittsburgh, Pa., a corpor was heated with 5.04 millimoles of BOs at 800° C. for ration of Pennsylvania 10 minutes. Boron trichloride and sulfur dioxide were O obtained in the volatile products, and a yellow solid, be No Drawing. Filed Mar. 28, 1958, Ser. No. 725,471 lieved to be sulfur, formed in the tube. 2 Claims. (CI. 23-205) Example IV.-9.32 millimoles of PC were passed over excess BO heated to 800° C. for 10 minutes. The vola This invention relates to the preparation of boron tri 15 tile products formed were analyzed with an infrared spec chloride and more particularly to the preparation of trometer and found to be predominantly BCl3. Some boron trichloride from boric oxide and non-metallic orange colored solids also formed in the reactor during chlorides. the reaction. The non-metallic chlorides which have been found Boron trichloride, BC, is used in several processes to useful in the practice of this invention are all volatile prepare other boron compounds, as a catalyst, and, in 20 liquids, at ordinary temperatures, and their reaction with general, is regarded as a basic boron compound. -

Boron and Titanium(IV) Halide Mediated Reactions
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Doctoral Dissertations Graduate School 8-2010 Boron and Titanium(IV) Halide Mediated Reactions Michael Patrick Quinn University of Tennessee - Knoxville, [email protected] Follow this and additional works at: https://trace.tennessee.edu/utk_graddiss Part of the Organic Chemistry Commons Recommended Citation Quinn, Michael Patrick, "Boron and Titanium(IV) Halide Mediated Reactions. " PhD diss., University of Tennessee, 2010. https://trace.tennessee.edu/utk_graddiss/908 This Dissertation is brought to you for free and open access by the Graduate School at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a dissertation written by Michael Patrick Quinn entitled "Boron and Titanium(IV) Halide Mediated Reactions." I have examined the final electronic copy of this dissertation for form and content and recommend that it be accepted in partial fulfillment of the requirements for the degree of Doctor of Philosophy, with a major in Chemistry. George W. Kabalka, Major Professor We have read this dissertation and recommend its acceptance: Shane Foister, Ziling (Ben) Xue, Paul Dalhaimer Accepted for the Council: Carolyn R. Hodges Vice Provost and Dean of the Graduate School (Original signatures are on file with official studentecor r ds.) To the Graduate Council: I am submitting herewith a dissertation written by Michael Patrick Quinn entitled ―Boron and Titanium(IV) Halide Mediated Reactions.‖ I have examined the final electronic copy of this dissertation for form and content and recommend that it be accepted in partial fulfillment of the requirements for the degree of Doctor of Philosophy, with a major in Chemistry. -

Equations of State of New Boron-Rich Selenides B6se and B12se
Equations of state of new boron-rich selenides B6Se and B12Se Kirill A. Cherednichenko,1,2 Yann Le Godec,3 and Vladimir L. Solozhenko 1,* 1 LSPM–CNRS, Université Sorbonne Paris Nord, 93430 Villetaneuse, France 2 Department of Physical and Colloid Chemistry, Gubkin University, Moscow, 119991, Russia 3 IMPMC–CNRS, UPMC Sorbonne Universités, 75005 Paris, France Abstract Two novel of boron-rich selenides, orthorhombic B6Se and rhombohedral B12Se, have been recently synthesized at high pressure – high temperature conditions. Room-temperature compressibilities of these phases were studied in a diamond anvil cell using synchrotron powder X-ray diffraction. A fit of experimental p-V data by third-order Birch-Murnaghan equation of state yielded the bulk moduli of 155(2) GPa for B12Se and 144(3) GPa for B6Se. No pressure-induced phase transitions have been observed in the studied pressure range, i.e. up to 35 GPa. Keywords : boron-rich selenides; high pressure; equation of state Introduction Boron-rich compounds containing B12-icosahedra have been known since 1960-s [1]. The majority of these compounds are the structural derivatives of elemental α-rhombohedral boron (α-B12) with general stoichiometry: B12Xy (where X is interstitial atom, y ≤ 2). They received a considerable attention due to the high hardness, chemical inertness, thermal conductivity, radiation resistance and unusual electronic properties [2-5]. For instance, boron carbide B12C3 is hard compound widely used as abrasive and armor material [6,7], boron suboxide B12O2 is the hardest known oxide [8,9], and B12As2 was found to be extremely stable under electron bombardment and able to the self-healing [10]. -

123. Antimony
1998:11 The Nordic Expert Group for Criteria Documentation of Health Risks from Chemicals 123. Antimony John Erik Berg Knut Skyberg Nordic Council of Ministers arbete och hälsa vetenskaplig skriftserie ISBN 91–7045–471–x ISSN 0346–7821 http://www.niwl.se/ah/ah.htm National Institute for Working Life National Institute for Working Life The National Institute for Working Life is Sweden's center for research and development on labour market, working life and work environment. Diffusion of infor- mation, training and teaching, local development and international collaboration are other important issues for the Institute. The R&D competence will be found in the following areas: Labour market and labour legislation, work organization and production technology, psychosocial working conditions, occupational medicine, allergy, effects on the nervous system, ergonomics, work environment technology and musculoskeletal disorders, chemical hazards and toxicology. A total of about 470 people work at the Institute, around 370 with research and development. The Institute’s staff includes 32 professors and in total 122 persons with a postdoctoral degree. The National Institute for Working Life has a large international collaboration in R&D, including a number of projects within the EC Framework Programme for Research and Technology Development. ARBETE OCH HÄLSA Redaktör: Anders Kjellberg Redaktionskommitté: Anders Colmsjö och Ewa Wigaeus Hjelm © Arbetslivsinstitutet & författarna 1998 Arbetslivsinstitutet, 171 84 Solna, Sverige ISBN 91–7045–471–X ISSN 0346-7821 Tryckt hos CM Gruppen Preface The Nordic Council is an intergovernmental collaborative body for the five countries, Denmark, Finland, Iceland, Norway and Sweden. One of the committees, the Nordic Senior Executive Committee for Occupational Environmental Matters, initiated a project in order to produce criteria documents to be used by the regulatory authorities in the Nordic countries as a scientific basis for the setting of national occupational exposure limits. -

Safety Data Sheet
SAFETY DATA SHEET 1. Identification Product identifier Boron Bromide (BBr3) Other means of identification SDS number 1EC Materion Code 1EC CAS number 10294-33-4 Synonyms BORON BROMIDE Manufacturer/Importer/Supplier/Distributor information Manufacturer Company name Materion Advanced Chemicals Inc. Address 407 N 13th Street 1316 W. St. Paul Avenue Milwaukee, WI 53233 United States Telephone 414.212.0257 E-mail [email protected] Contact person Noreen Atkinson Emergency phone number Chemtrec 800.424.9300 2. Hazard(s) identification Physical hazards Not classified. Health hazards Acute toxicity, oral Category 2 Acute toxicity, inhalation Category 1 Skin corrosion/irritation Category 1A Serious eye damage/eye irritation Category 1 Environmental hazards Not classified. OSHA defined hazards Not classified. Label elements Signal word Danger Hazard statement Fatal if swallowed. Causes severe skin burns and eye damage. Causes serious eye damage. Fatal if inhaled. Precautionary statement Prevention Do not breathe vapor. Wash thoroughly after handling. Do not eat, drink or smoke when using this product. Use only outdoors or in a well-ventilated area. Wear protective gloves/protective clothing/eye protection/face protection. Wear respiratory protection. Response If swallowed: Immediately call a poison center/doctor. If swallowed: Rinse mouth. Do NOT induce vomiting. If on skin (or hair): Take off immediately all contaminated clothing. Rinse skin with water/shower. If inhaled: Remove person to fresh air and keep comfortable for breathing. If in eyes: Rinse cautiously with water for several minutes. Remove contact lenses, if present and easy to do. Continue rinsing. Immediately call a poison center/doctor. Specific treatment is urgent (see this label). -

United States Patent Office Patented Sept
2,904,588 United States Patent Office Patented Sept. 15, 1959 - 2 m Three grams of the product prepared as described above was placed in a polyethylene bottle and 6 g. of water 2,904,588 was added. After the initial exothermic reaction had FLUOROPHOSPHORANES AND THEIR subsided and the solution had cooled, a white crystalline PREPARATION solid separated. This solid was recrystallized twice from water. After drying, the product melted at 159 to 161 William C. Smith, Wilmington, Del, assignor to E. I. du C. The melting point of benzenephosphonic acid is 159 Pont de Nemours and Company, Wilmington, Del., a to 161° C., and this is the product expected from the com corporation of Delaware plete hydrolysis of phenyltetrafluorophosphorane. No Drawing. Application March 26, 1956 O Nuclear magnetic resonance examination of the phenyl Serial No. 573,659 tetrafluorophosphorane showed it to have four equiv alent fluorine atoms bound to phosphorus, which indi 15 Claims. (C. 260-543) cated a square pyramidal structure. This invention relates to new compositions of matter 5 EXAMPLE II and to their preparation. Example I was repeated, using a charge consisting Organic fluorine compounds have attained considerable of 53.7 g (0.3 mole) of phenylphosphonous dichloride importance in recent years and simple and economic in the reaction flask and 65.1 g (0.3 mole) of antimony methods for their preparation are greatly desired. pentafluoride in the dropping funnel. The antimony This invention has as an object the preparation of new 20 pentafluoride was added to the phenylphosphonous di fluorophosphoranes. A further object is the provision of chloride at such a rate that the temperature of the reac a new process for their preparation.