Towards Electron Encapsulation. Polynitrile Approach. Abstract
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
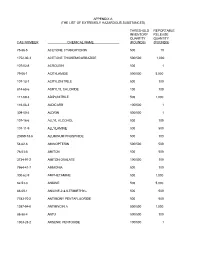
The List of Extremely Hazardous Substances)
APPENDIX A (THE LIST OF EXTREMELY HAZARDOUS SUBSTANCES) THRESHOLD REPORTABLE INVENTORY RELEASE QUANTITY QUANTITY CAS NUMBER CHEMICAL NAME (POUNDS) (POUNDS) 75-86-5 ACETONE CYANOHYDRIN 500 10 1752-30-3 ACETONE THIOSEMICARBAZIDE 500/500 1,000 107-02-8 ACROLEIN 500 1 79-06-1 ACRYLAMIDE 500/500 5,000 107-13-1 ACRYLONITRILE 500 100 814-68-6 ACRYLYL CHLORIDE 100 100 111-69-3 ADIPONITRILE 500 1,000 116-06-3 ALDICARB 100/500 1 309-00-2 ALDRIN 500/500 1 107-18-6 ALLYL ALCOHOL 500 100 107-11-9 ALLYLAMINE 500 500 20859-73-8 ALUMINUM PHOSPHIDE 500 100 54-62-6 AMINOPTERIN 500/500 500 78-53-5 AMITON 500 500 3734-97-2 AMITON OXALATE 100/500 100 7664-41-7 AMMONIA 500 100 300-62-9 AMPHETAMINE 500 1,000 62-53-3 ANILINE 500 5,000 88-05-1 ANILINE,2,4,6-TRIMETHYL- 500 500 7783-70-2 ANTIMONY PENTAFLUORIDE 500 500 1397-94-0 ANTIMYCIN A 500/500 1,000 86-88-4 ANTU 500/500 100 1303-28-2 ARSENIC PENTOXIDE 100/500 1 THRESHOLD REPORTABLE INVENTORY RELEASE QUANTITY QUANTITY CAS NUMBER CHEMICAL NAME (POUNDS) (POUNDS) 1327-53-3 ARSENOUS OXIDE 100/500 1 7784-34-1 ARSENOUS TRICHLORIDE 500 1 7784-42-1 ARSINE 100 100 2642-71-9 AZINPHOS-ETHYL 100/500 100 86-50-0 AZINPHOS-METHYL 10/500 1 98-87-3 BENZAL CHLORIDE 500 5,000 98-16-8 BENZENAMINE, 3-(TRIFLUOROMETHYL)- 500 500 100-14-1 BENZENE, 1-(CHLOROMETHYL)-4-NITRO- 500/500 500 98-05-5 BENZENEARSONIC ACID 10/500 10 3615-21-2 BENZIMIDAZOLE, 4,5-DICHLORO-2-(TRI- 500/500 500 FLUOROMETHYL)- 98-07-7 BENZOTRICHLORIDE 100 10 100-44-7 BENZYL CHLORIDE 500 100 140-29-4 BENZYL CYANIDE 500 500 15271-41-7 BICYCLO[2.2.1]HEPTANE-2-CARBONITRILE,5- -

Scientific Advisory Board
OPCW Scientific Advisory Board Eleventh Session SAB-11/1 11 – 13 February 2008 13 February 2008 Original: ENGLISH REPORT OF THE ELEVENTH SESSION OF THE SCIENTIFIC ADVISORY BOARD 1. AGENDA ITEM ONE – Opening of the Session The Scientific Advisory Board (SAB) met for its Eleventh Session from 11 to 13 February 2008 at the OPCW headquarters in The Hague, the Netherlands. The Session was opened by the Vice-Chairperson of the SAB, Mahdi Balali-Mood. The meeting was chaired by Philip Coleman of South Africa, and Mahdi Balali-Mood of the Islamic Republic of Iran served as Vice-Chairperson. A list of participants appears as Annex 1 to this report. 2. AGENDA ITEM TWO – Adoption of the agenda 2.1 The SAB adopted the following agenda for its Eleventh Session: 1. Opening of the Session 2. Adoption of the agenda 3. Tour de table to introduce new SAB Members 4. Election of the Chairperson and the Vice-Chairperson of the SAB1 5. Welcome address by the Director-General 6. Overview on developments at the OPCW since the last session of the SAB 7. Establishment of a drafting committee 8. Work of the temporary working groups: (a) Consideration of the report of the second meeting of the sampling-and-analysis temporary working group; 1 In accordance with paragraph 1.1 of the rules of procedure for the SAB and the temporary working groups of scientific experts (EC-XIII/DG.2, dated 20 October 1998) CS-2008-5438(E) distributed 28/02/2008 *CS-2008-5438.E* SAB-11/1 page 2 (b) Status report by the Industry Verification Branch on the implementation of sampling and analysis for Article VI inspections; (c) Presentation by the OPCW Laboratory; (d) Update on education and outreach; and (e) Update on the formation of the temporary working group on advances in science and technology and their potential impact on the implementation of the Convention: (i) composition of the group; and (ii) its terms of reference 9. -

House Fly Attractants and Arrestante: Screening of Chemicals Possessing Cyanide, Thiocyanate, Or Isothiocyanate Radicals
House Fly Attractants and Arrestante: Screening of Chemicals Possessing Cyanide, Thiocyanate, or Isothiocyanate Radicals Agriculture Handbook No. 403 Agricultural Research Service UNITED STATES DEPARTMENT OF AGRICULTURE Contents Page Methods 1 Results and discussion 3 Thiocyanic acid esters 8 Straight-chain nitriles 10 Propionitrile derivatives 10 Conclusions 24 Summary 25 Literature cited 26 This publication reports research involving pesticides. It does not contain recommendations for their use, nor does it imply that the uses discussed here have been registered. All uses of pesticides must be registered by appropriate State and Federal agencies before they can be recommended. CAUTION: Pesticides can be injurious to humans, domestic animals, desirable plants, and fish or other wildlife—if they are not handled or applied properly. Use all pesticides selectively and carefully. Follow recommended practices for the disposal of surplus pesticides and pesticide containers. ¿/áepé4áaUÁí^a¡eé —' ■ -"" TMK LABIL Mention of a proprietary product in this publication does not constitute a guarantee or warranty by the U.S. Department of Agriculture over other products not mentioned. Washington, D.C. Issued July 1971 For sale by the Superintendent of Documents, U.S. Government Printing Office Washington, D.C. 20402 - Price 25 cents House Fly Attractants and Arrestants: Screening of Chemicals Possessing Cyanide, Thiocyanate, or Isothiocyanate Radicals BY M. S. MAYER, Entomology Research Division, Agricultural Research Service ^ Few chemicals possessing cyanide (-CN), thio- cyanate was slightly attractive to Musca domes- eyanate (-SCN), or isothiocyanate (~NCS) radi- tica, but it was considered to be one of the better cals have been tested as attractants for the house repellents for Phormia regina (Meigen). -

IUPAC Provisional Recommendations
Preferred IUPAC Names 177 Chapter 6, Sect 66 September, 2004 P-66 Amides, imides, hydrazides, nitriles, aldehydes, their chalcogen analogues and derivatives P-66.0 Introduction P-66.1 Amides P-66.2 Imides P-66.3 Hydrazides P-66.4 Amidines, amidrazones, hydrazidines P-66.5 Nitriles P-66.6 Aldehydes P-66.0 Introduction The classes dealt with in this Section have in common the fact that their retained names are derived from those of acids by changing the ‘ic acid’ ending to a class name, for example ‘amide’, ‘ohydrazide’, ‘nitrile’ or ‘aldehyde’. Their systematic names are formed substitutively by the suffix mode using one of two types of suffix, one that includes the carbon atom, for example, ‘carbonitrile’ for −CN, and one that does not, for example, ‘-nitrile’ for −(C)N,. Amidines are named as amides, hydrazidines as hydrazides, and amidrazones as amides or hydrazides. P-66.1 Amides P-66.1.0 Introduction P-66.1.1 Primary amides P-66.1.2 Secondary and tertiary amides P-66.1.3 Chalcogen analogues of amides P-66.1.4 Lactams, lactims, sultams and sultims P-66.1.5 Amides of carbonic, oxalic, cyanic, and polycarbonic acids P-66.1.6 Polyfunctional amides P-66.1.0 Introduction Amides are derivatives of oxoacids in which each hydroxy group has been replaced by an amino or substituted amino group. Chalcogen replacement analogues are called thio-, seleno- and telluroamides. Compounds having one, two or three acyl groups on a single nitrogen atom are generically included and may be designated as primary, secondary and tertiary amides, respectively. -
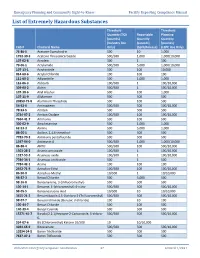
List of Extremely Hazardous Substances
Emergency Planning and Community Right-to-Know Facility Reporting Compliance Manual List of Extremely Hazardous Substances Threshold Threshold Quantity (TQ) Reportable Planning (pounds) Quantity Quantity (Industry Use (pounds) (pounds) CAS # Chemical Name Only) (Spill/Release) (LEPC Use Only) 75-86-5 Acetone Cyanohydrin 500 10 1,000 1752-30-3 Acetone Thiosemicarbazide 500/500 1,000 1,000/10,000 107-02-8 Acrolein 500 1 500 79-06-1 Acrylamide 500/500 5,000 1,000/10,000 107-13-1 Acrylonitrile 500 100 10,000 814-68-6 Acrylyl Chloride 100 100 100 111-69-3 Adiponitrile 500 1,000 1,000 116-06-3 Aldicarb 100/500 1 100/10,000 309-00-2 Aldrin 500/500 1 500/10,000 107-18-6 Allyl Alcohol 500 100 1,000 107-11-9 Allylamine 500 500 500 20859-73-8 Aluminum Phosphide 500 100 500 54-62-6 Aminopterin 500/500 500 500/10,000 78-53-5 Amiton 500 500 500 3734-97-2 Amiton Oxalate 100/500 100 100/10,000 7664-41-7 Ammonia 500 100 500 300-62-9 Amphetamine 500 1,000 1,000 62-53-3 Aniline 500 5,000 1,000 88-05-1 Aniline, 2,4,6-trimethyl- 500 500 500 7783-70-2 Antimony pentafluoride 500 500 500 1397-94-0 Antimycin A 500/500 1,000 1,000/10,000 86-88-4 ANTU 500/500 100 500/10,000 1303-28-2 Arsenic pentoxide 100/500 1 100/10,000 1327-53-3 Arsenous oxide 100/500 1 100/10,000 7784-34-1 Arsenous trichloride 500 1 500 7784-42-1 Arsine 100 100 100 2642-71-9 Azinphos-Ethyl 100/500 100 100/10,000 86-50-0 Azinphos-Methyl 10/500 1 10/10,000 98-87-3 Benzal Chloride 500 5,000 500 98-16-8 Benzenamine, 3-(trifluoromethyl)- 500 500 500 100-14-1 Benzene, 1-(chloromethyl)-4-nitro- 500/500 -

565.Full.Pdf
Studies on the Biological Action of Malononitriles* I. The Effect of Substituted Malononitriles on the Growth of Transplanted Tumors in Mice EMERYM. GAL,| FUNG-ÜAANFUNG,ANDDAVIDM. GREENBERG ASSISTEDBYHARRIETEZRAANDSHERBURNEF.COOK,JB. (Department of Biochemistry, School of Medicine, Unirersity of California, Berkeley, Calif.') This laboratory has been engaged for a number MATERIALS of years on a program of cancer chemotherapy re Malononitrile, by virtue of its active méthylènegroup, search guided mainly by the concept of metabolite readily condenses with carbonyl compounds to yield dinitriles. The condensation products show different degrees of saturation antagonism (9) in the selection of compounds to be and can be divided into the following groups: tested for chemotherapeutic activity. The impor 1. R-CH,CH(CN), tance attributed to the components of the nucleic 2. R-CH (CH[CN]S), acids in the genesis of neoplasms and the control of 3. R-CH:C(CN)j cellular functions has, with some measure of suc R.-. cess, oriented extensive investigations on experi 4. R2-C:C(CN)S 5. R-N:N-CH(CN)2 mental tumor chemotherapy. An outstanding ex ample is the work of Kidder and his associates (13) With a few exceptions the compounds were prepared in our on the growth-inhibiting properties of the purine laboratory according to known procedures (1, 4, 14). The car analog, 8-azaguanine. bonyl compounds used in the syntheses were, for the most part, purchased from Distillation Products Industries. The prepared Our efforts have been considerably influenced compounds were checked for purity and the elementary compo by the work of Hyden and Hartelius (12), who re sition of those which were not described in the literature deter ported that administration of malononitrile mined by micro-analysis. -

List of Lists
United States Office of Solid Waste EPA 550-B-10-001 Environmental Protection and Emergency Response May 2010 Agency www.epa.gov/emergencies LIST OF LISTS Consolidated List of Chemicals Subject to the Emergency Planning and Community Right- To-Know Act (EPCRA), Comprehensive Environmental Response, Compensation and Liability Act (CERCLA) and Section 112(r) of the Clean Air Act • EPCRA Section 302 Extremely Hazardous Substances • CERCLA Hazardous Substances • EPCRA Section 313 Toxic Chemicals • CAA 112(r) Regulated Chemicals For Accidental Release Prevention Office of Emergency Management This page intentionally left blank. TABLE OF CONTENTS Page Introduction................................................................................................................................................ i List of Lists – Conslidated List of Chemicals (by CAS #) Subject to the Emergency Planning and Community Right-to-Know Act (EPCRA), Comprehensive Environmental Response, Compensation and Liability Act (CERCLA) and Section 112(r) of the Clean Air Act ................................................. 1 Appendix A: Alphabetical Listing of Consolidated List ..................................................................... A-1 Appendix B: Radionuclides Listed Under CERCLA .......................................................................... B-1 Appendix C: RCRA Waste Streams and Unlisted Hazardous Wastes................................................ C-1 This page intentionally left blank. LIST OF LISTS Consolidated List of Chemicals -

Polymers Functionalized with Nitrile Compounds
(19) TZZ ¥ Z_T (11) EP 2 382 240 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.: of the grant of the patent: C08C 19/44 (2006.01) B60C 1/00 (2006.01) 16.04.2014 Bulletin 2014/16 C08K 3/00 (2006.01) C08L 15/00 (2006.01) (21) Application number: 10733884.0 (86) International application number: PCT/US2010/021767 (22) Date of filing: 22.01.2010 (87) International publication number: WO 2010/085622 (29.07.2010 Gazette 2010/30) (54) POLYMERS FUNCTIONALIZED WITH NITRILE COMPOUNDS CONTAINING A PROTECTED AMINO GROUP DURCH NITRILVERBINDUNGEN MIT EINER GESCHÜTZTEN AMINOGRUPPE FUNKTIONALISIERTE POLYMERE POLYMÈRES FONCTIONNALISÉS AVEC DES COMPOSÉS DE NITRILE CONTENANT UN GROUPE AMINO PROTÉGÉ (84) Designated Contracting States: • SUZUKI, Eiju AT BE BG CH CY CZ DE DK EE ES FI FR GB GR Tokyo 104-8340 (JP) HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL • BRUMBAUGH, Dennis, R. PT RO SE SI SK SM TR North Canton, OH 44721 (US) (30) Priority: 23.01.2009 US 146871 P (74) Representative: Waldren, Robin Michael et al Marks & Clerk LLP (43) Date of publication of application: 90 Long Acre 02.11.2011 Bulletin 2011/44 London WC2E 9RA (GB) (73) Proprietor: Bridgestone Corporation Tokyo 104-8340 (JP) (56) References cited: WO-A1-2006/128158 WO-A2-2008/156788 (72) Inventors: KR-A- 20070 092 699 US-A- 4 927 887 • LUO, Steven US-A- 5 310 798 US-A1- 2002 137 849 Copley, OH 44321 (US) US-A1- 2003 176 276 US-B2- 6 897 270 Note: Within nine months of the publication of the mention of the grant of the European patent in the European Patent Bulletin, any person may give notice to the European Patent Office of opposition to that patent, in accordance with the Implementing Regulations. -
Acetone Cyanohydrin Final AEGL Document
Acute Exposure Guidelines for Selected Airborne Chemicals, Volume 7 http://www.nap.edu/catalog/12503.html Committee on Acute Exposure Guideline Levels Committee on Toxicology Board on Environmental Studies and Toxicology Division on Earth and Life Studies Copyright © National Academy of Sciences. All rights reserved. Acute Exposure Guidelines for Selected Airborne Chemicals, Volume 7 http://www.nap.edu/catalog/12503.html THE NATIONAL ACADEMIES PRESS 500 FIFTH STREET, NW WASHINGTON, DC 20001 NOTICE: The project that is the subject of this report was approved by the Governing Board of the National Research Council, whose members are drawn from the councils of the National Academy of Sciences, the National Academy of Engineering, and the Institute of Medicine. The members of the committee responsible for the report were chosen for their special competences and with regard for appropriate balance. This project was supported by Contract No. W81K04-06-D-0023 between the National Academy of Sciences and the U.S. Department of Defense. Any opinions, findings, conclu- sions, or recommendations expressed in this publication are those of the author(s) and do not necessarily reflect the view of the organizations or agencies that provided support for this project. International Standard Book Number-13: 978-0-309-12755-4 International Standard Book Number-10: 0-309-12755-6 Additional copies of this report are available from The National Academies Press 500 Fifth Street, NW Box 285 Washington, DC 20055 800-624-6242 202-334-3313 (in the Washington metropolitan area) http://www.nap.edu Copyright 2009 by the National Academy of Sciences. All rights reserved. -
Electrochemical Analyses and Reactions of Malononitrile Derivatives and Phenyl Azide a Research Paper Submitted to the Graduate
ELECTROCHEMICAL ANALYSES AND REACTIONS OF MALONONITRILE DERIVATIVES AND PHENYL AZIDE A RESEARCH PAPER SUBMITTED TO THE GRADUATE SCHOOL IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE MASTERS OF ART BY JINYU LIU DR. CHONG ‐ ADVISOR BALL STATE UNIVERSITY MUNCIE, INDIANA JULY 2013 CONTENT Abstract 1 Introduction 1 Material 3 4-methoxybenzalmalonoitrile 3 Phenyl azide: 4 Set up 6 Experiment 6 1. 4-methoxybenzalmalonoitrile 6 a. Low concentration experiment to check and determine the potential of 4-methoxybenzalmalononitrile. 6 b. High concentration experiment to confirm the reaction and the product. 8 c. Product isolation. 10 2. Electronic effect for different substituted group 12 3. Phenyl azide reaction. 15 Result 16 Acknowledgement 18 Reference 19 Abstract: The versatile and relatively inexpensive (low energy cost) synthesis of new organic molecules is important for pharmaceutical production, polymerization, and other industrially applicable facets of chemistry. Electrochemical reduction of unsaturated organic compounds at room temperature to new organic products offers a benign and ambient method for the production of such molecules. The compounds, (p-methoxybenzal)malononitrile, 1 , Epc = 0/+ -1.64 V vs. Ferrocene/Ferrocenium (Cp2Fe ), its derivatives and phenyl azide, 2 , Epc = 0/+ -2.43 V vs. Cp2Fe , were investigated for their redox activities with and without alkylation agents, R-X, via scanning and pulse voltammetric techniques in acetonitrile, acetonitrile/water and tetrahydrofuran as solvents, using 0.1 M [NBu4][PF6] as the supporting electrolyte. The redox potentials were measured at glassy carbon and platinum disk 0/+ electrodes. Controlled potential electrolysis (CPE) of 1 at Eappl = -1.75 V vs. Cp2Fe was 0/+ yielding p-methoxybenzaldehyde (> 90%). -
The Synthesis and Characterisation of Some Organic
The Synthesis And Characterisation Of Some Organic Dicyanomethylene Salts A thesis presented for the degree of M.Sc. by Orla Wilson BSc (Hons) at DUBLIN CITY UNIVERSITY School of Chemical Sciences November 1997 For my family Una, Sam, Fiona and Kathryn ii Declaration I, the undersigned, hereby declare that this thesis, which I now submit for assessment on the programme of study leading to the award of M.Sc. represents the sole work of the author and has not been taken from the work of others save and to the extent that such work has been cited and acknowledged within the text. Orla Wilson Acknowledgements I would like to thank my supervisor Prof. Albert Pratt for his help, encouragement and guidance during the course of this work. A huge thank you to the academic staff in general and in particular to the technical staff of the School of Chemical Sciences for their constant help and humour along the way. I want to thank Mick Burke for all his help, also Maurice, Damien, Veronica, Anne and the rest of the technicians. I want to say the heartiest of thanks to Siobh and Ciara B who were with me in AG07 and also Ciara H, Monica, Susan, Davnet and Cyril who were there in spirit - thanks for all the support. The members of the Albert Pratt research group past and present also deserve thanks - namely Ben, Colette, Colm, Cormac, Dawn, Farmer, James, Joe, Mark, Mauro, Ollie, Owain, Rod and Shane - as well as the other occupants of AG07, next door and upstairs over the years - Mary, Charlie, Deirdre, Bronagh, Karen, Frances, Rachel, Noel, Christine, Una, Luke, Ciaran, Mikey, Padraig, Paul, James and Kevin. -

Selected Specific Rates of Reactions of Transients from Water in Aqueous
1 Allioa 1 4 5 7 5 ^ NA1 L IN®T STANDARDS & ii i TECH R.I.C. All 102145759 /NSRDS-NBS 1964 PLICATIONS NSRDS-NBS 51 * *°*tAU Of U.S. DEPARTMENT OF COMMERCE / National Bureau of Standards (Site N SRP S Selected Specific Rates of Reactions of Transients from Water in Aqueous Solution. II. Hydrogen Atom too 513 \o.5 <t15 Hi NATIONAL BUREAU OF STANDARDS 1 The National Bureau of Standards was established by an act of Congress March 3, 1901. The Bureau’s overall goal is to strengthen and advance the Nation’s science and technology and facilitate their effective application for public benefit. To this end, the Bureau conducts research and provides: (1) a basis for the Nation's physical measurement system, (2) scientific and technological services for industry and government, (3) a technical basis for equity in trade, and (4) technical services to promote public safety. The Bureau consists of the Institute for Basic Standards, the Institute for Materials Research, the Institute for Applied Technology, the Institute for Computer Sciences and Technology, and the Office for Information Programs. THE INSTITUTE FOR BASIC STANDARDS provides the central basis within the United States of a complete and consistent system of physical measurement; coordinates that system with measurement systems of other nations; and furnishes essential services leading to accurate and uniform physical measurements throughout the Nation’s scientific community, industry, and commerce. The Institute consists of a Center for Radiation Research, an Office of Meas- urement Services and the following divisions: Applied Mathematics — Electricity — Mechanics — Heat — Optical Physics — Nuclear Sciences 2 — Applied Radiation 2 — Quantum Electronics 3 — Electromagnetics 3 — Time 3 3 3 and Frequency — Laboratory Astrophysics — Cryogenics .