The Synthesis and Characterisation of Some Organic
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

2-Adamantylacetic Acid* B
CROATICA CHEM1CA ACTA CcACAA 48 (2) I69-i78 (1976) YU ISSN 0011-1643 CCA-927 547.466:542.91 Originai Scientific Paper Synthesis of a-Amino-1-Adamantylacetic and a-Amino -2-Adamantylacetic Acid* B . Gaspert, S. Hromadko, and B. Vranesic Research Department »Piiva«, Pharmaceuticai and Chemicai Works, Zagreb, Croatia, Yugosiavia Rece ived September 29 , 1975 a-Amino-2-adamantylacetic acid (VII) was obtained when 2-adamantylcyanoacetylhydrazide (IX) was subjected to Curtius rearrangement or 2-(2-adaman;tyl)-malonamic acid (XII) to Hof mann degradation. The same amino acid was obtained when ri. -bromo-2-adamantylacetic acid (VI) was •treated with ammonia. Partial hydrolysis of diethyl adamantyl-(1)-malonate (XIV) yie1ded ethyl adamantyl-(1)-malcmate (XV) which, after treatment with thionyl chlodde and then ammonia, gave 2-(1-adamantyl) -malonamic acid ethyl ester (XVII). Hofmann degradation of 2-(1 -adamantyl)-malonamic acid ethyl ester gave a-amino-1- -adama:ntylacetic acid (XVIII). Discovery of interesting biological properties of 1-aminoadamantane hy drochloride' ·stimulated the synthesis of a large number of structurally related compounds. The synthesis of amino acids, having an adamantyl group as a part of the 2 molecule, has been reported for 3-aminoadamantane-1-carboxyHc acid , a -amino-1-adamantylacetic acid3 and recently for 2-'aminoadamantane-2-car boxylic acid4. It seemed appmpriate to us to synthetise a-amLno-2-adamantyl acetic acid5 and to elaborate a moire convenient synthesis of a-amino-1-ada ma:ntylacetic acid6, which -

Synthesis of 3-Fluorooxindole Derivatives Using Diethyl 2-Fluoromalonate
Durham E-Theses Synthesis of 3-uoro-oxindoles and phenyl uoroacetic acid derivatives HARSANYI, ANTAL How to cite: HARSANYI, ANTAL (2013) Synthesis of 3-uoro-oxindoles and phenyl uoroacetic acid derivatives, Durham theses, Durham University. Available at Durham E-Theses Online: http://etheses.dur.ac.uk/6357/ Use policy The full-text may be used and/or reproduced, and given to third parties in any format or medium, without prior permission or charge, for personal research or study, educational, or not-for-prot purposes provided that: • a full bibliographic reference is made to the original source • a link is made to the metadata record in Durham E-Theses • the full-text is not changed in any way The full-text must not be sold in any format or medium without the formal permission of the copyright holders. Please consult the full Durham E-Theses policy for further details. Academic Support Oce, Durham University, University Oce, Old Elvet, Durham DH1 3HP e-mail: [email protected] Tel: +44 0191 334 6107 http://etheses.dur.ac.uk A Thesis Entitled Synthesis of 3-fluoro-oxindoles and phenyl fluoroacetic acid derivatives Submitted by Antal Harsanyi BSc (College of St. Hild and St. Bede) Department of Chemistry A Candidate for the Degree of Master of Science 2012 Declaration The work presented within this thesis was carried out at Durham University between October 2011 and August 2012. This thesis is the work of the author, except where acknowledged by reference and has not been submitted for any other degree. Part of this work has been presented at: 12th RSC Annual Fluorine Group Meeting, St Andrews, August 2012 Statement of copyright The copyright of this thesis rests with the author. -

The Development and Application of Metal-Catalyzed Processes for Organic Synthesis
The Development and Application of Metal-Catalyzed Processes for Organic Synthesis by Edward J. Hennessy B.A. Chemistry Northwestern University, 2000 Submitted to the Department of Chemistry in Partial Fulfillment of the Requirements for the Degree of DOCTOR OF PHILOSOPHY IN ORGANIC CHEMISTRY at the Massachusetts Institute of Technology June 2005 © 2005 Massachusetts Institute of Technology All Rights Reserved Signature of Author: " - - 1_"' .Da-uirnt of Chemistry May 19, 2005 Certified by: Stephen L. Buchwald Camille Dreyfus Professor of Chemistry Thesis Supervisor Accepted by: Robert W. Field Chairman, Departmental Committee on Graduate Students ARCHIVES: This doctoral thesis has been examined by a committee of the Department of Chemistry as follows: -/ Professor Gregory C. Fu: " Ch Chair Professor Stephen L. Buchwald Thesis Supervisor Professor Timothy M. Swager: , -L i \ O 2 The Development and Application of Metal-Catalyzed Processes for Organic Synthesis by Edward J. Hennessy Submitted to the Department of Chemistry on May 19, 2005 in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy at the Massachusetts Institute of Technology ABSTRACT Chapter 1. Copper-Catalyzed Arylation of Stabilized Carbanions A mild, general catalytic system for the synthesis of a-aryl malonates has been developed. Aryl iodides bearing a variety of functional groups can be effectively coupled to diethyl malonate in high yields using inexpensive and widely available reagents, making this a superior method to those previously described that employ copper reagents or catalysts. The functional group tolerance of the process developed makes it complementary to analogous palladium-catalyzed couplings. Importantly, a set of mild reaction conditions has been developed that minimize product decomposition, a problem that had not been addressed previously in the literature. -

Knoevenagel Condensation
Knoevenagel condensation The Knoevenagel condensation (pronounced [ˈknøːvənaːɡl̩ ]) Knoevenagel condensation reaction is an organic reaction named after Emil Knoevenagel. It is a Named after Emil Knoevenagel modification of the aldol condensation.[1][2] Reaction type Coupling reaction A Knoevenagel condensation is a nucleophilic addition of an active Identifiers hydrogen compound to a carbonyl group followed by a dehydration reaction in which a molecule of water is eliminated (hence Organic knoevenagel- condensation). The product is often an α,β-unsaturated ketone (a Chemistry condensation conjugated enone). Portal RSC ontology RXNO:0000044 ID In this reaction the carbonyl group is an aldehyde or a ketone. The catalyst is usually a weakly basic amine. The active hydrogen component has the form[3] Z–CH2-Z or Z–CHR–Z for instance diethyl malonate, Meldrum's acid, ethyl acetoacetate or malonic acid, or cyanoacetic acid.[4] Z–CHR1R2 for instance nitromethane. where Z is an electron withdrawing functional group. Z must be powerful enough to facilitate deprotonation to the enolate ion even with a mild base. Using a strong base in this reaction would induce self-condensation of the aldehyde or ketone. The Hantzsch pyridine synthesis, the Gewald reaction and the Feist–Benary furan synthesis all contain a Knoevenagel reaction step. The reaction also led to the discovery of CS gas. Contents Doebner modification Scope Weiss–Cook reaction See also References Doebner modification The Doebner modification of the Knoevenagel condensation. Acrolein and malonic acid react in pyridine to give trans-2,4-pentadienoic acid with the loss of carbon dioxide. With malonic compounds the reaction product can lose a molecule of carbon dioxide in a subsequent step. -

Design, Synthesis, and Supramolecular Surface Chemistry Of
DESIGN, SYNTHESIS, AND SUPRAMOLECULAR SURFACE CHEMISTRY OF BI- AND TRIDENTATE SURFACE ANCHORS FOR NANOSCIENCE AND NANOBIOTECHNOLOGY A Dissertation Presented to The Graduate Faculty of the University of Akron In Partial Fulfillment Of the Requirements for the Degree Doctor of Philosophy Hui Wang August, 2007 DESIGN, SYNTHESIS, AND SUPRAMOLECULAR SURFACE CHEMISTRY OF BI- AND TRIDENTATE SURFACE ANCHORS FOR NANOSCIENCE AND NANOBIOTECHNOLOGY Hui Wang Dissertation Approved: Accepted: _______________________ _______________________ Advisor Department Chair Dr. Jun Hu Dr. Kim C. Calvo _______________________ _______________________ Committee Member Dean of the College Dr. Gerald F. Koser Dr. Ronald F. Levant _______________________ _______________________ Committee Member Dean of the Graduate School Dr. David A. Modarelli Dr. George R. Newkome _______________________ _______________________ Committee Member Date Dr. Claire A. Tessier _______________________ Committee Member Dr. Robert R. Mallik ii ABSTRACT This dissertation describes the design, synthesis, and supramolecular surface chemistry of bi- and tri-dentate surface anchors for nanoscience and nanobiotechnology. Molecular electronic device candidates, based on the tridentate surface anchor 2,4,9-trithia-tricyclo[3.3.1.13,7]decane, were used to bridge two ruthenium metal clusters. These well-designed ruthenium complexes were used as nanometer-sized molecular connector/metal cluster models to investigate the surface binding characteristics of tridentate surface anchor-metal junctions. Bi-dentate surface anchors, 1,4-dimercapto-2,3-dimethyl-butane- 2,3-diol and 4,5-dimethyl-2-(4-vinyl-phenyl)-[1,3,2]dioxaborolane-4,5-dithiol, were synthesized. They were used as ligands for stabilizing gold nanoparticles by two methods: direct reduction reaction, and ligand exchange reaction with triphenylphosphine-stabilized gold nanoparticles. -

Knoevenagel Condensation Reaction Catalysed by Agro-Waste Extract As a Greener Solvent Catalyst
ORIGINAL ARTICLE Org. Commun. 14:1 (2021) 81-91 Knoevenagel condensation reaction catalysed by agro-waste extract as a greener solvent catalyst Krishnappa B. Badiger and Kantharaju Kamanna * Peptide and Medicinal Chemistry Research Laboratory, Department of Chemistry, Rani Channamma University, P-B, NH-4, Belagavi 591 156, India (Received January 22, 2021; Revised March 15, 2021; Accepted March 18, 2021) Abstract: This paper present a novel Knoevenagel reaction protocol for the condensation of aromatic/heteroaromatic aldehydes with malononitrile to give α, β–unsaturated benzylidene derivatives. The main focus of this work is to reveal the usability of agro-waste extracts as a catalyst in the Knoevenagel condensation. The present protocol proceeds efficiently for various substituted aromatic and heterocyclic aldehydes in the Knoevenagel reactions. In addition, the present method describes direct isolation of the formed products without using organic solvent extraction gave good yields product. Keywords: one-pot reaction; green chemistry; solvent-free; Knoevenagel condensation reaction; malononitrile; room temperature. ©2021 ACG Publication. All right reserved. 1. Introduction The carbon-carbon bond formation via Knoevenagel condensation1-3 is one of the most important routes repeated in the synthetic organic chemistry and allows the production of various active pharmaceutical molecules.4 Figure 1. Structure of biologically potent benzylidinemalononitrile derivatives * Corresponding author:E-Mail: [email protected] The article was published -

Acetoacetic Ester Synthesis
Programme: B.Sc. B.ed. (Integrated) Course: ORGANIC CHEMISTRY- III Semester: VI Code: CHE-352 Topic: ETHYLACETOACETATEE Date- 07/04/2020 y Only PPe Dr. Angad Kumar Singh ForF Department of Chemistry, Central University of South Bihar, Gaya (Bihar) Note: These materials are only for classroom teaching purpose at Central University of South Bihar. All the data taken from several books, research articles including Wikipedia. Note: These materials are only for classroom teaching purpose at Central University of South Bihar. All the data taken from several books, research articles including Wikipedia. Ethylacetoacetate The Claisen Condensation between esters containing - hdhydrogens, promotdted byabase such as sodium ethoxy ide, toproduce a !- ketoester. One equiv serves as the nucleophilele (enolate)(eno and the other is OnlyO the electrophile which undergoes additiontion andae elimination. The use of stronger bases, e.g. sodium amide or sodiumsodUse hydride instead of sodium ethoxide, often increases the yield.d. al onal Ethylacetoacetate Note: These materials are only for classroom teaching purpose at Central University of South Bihar. All the data taken from several books, research articles including Wikipedia. Mechanism of the Claisen Condensation The reaction is driven to product by the final deprotonation step. Note: These materials are only for classroom teaching purpose at Central University of South Bihar. All the data taken from several books, research articles including Wikipedia. Mixed Claisen Condensation Like mixed aldol reactions, mixed Claisen condensations are useful if differences in reactivity exist betweenn they two esters as for example when one of the esters has no -hydrogenydrogOnlyO . Examples of such esters are: e Use ers excess Note: These materials are only for classroom teaching purpose at Central University of South Bihar. -

Diethyl Malonate
DIETHYL MALONATE PRODUCT IDENTIFICATION CAS NO. 105-53-3 EINECS NO. 203-305-9 FORMULA CH 2(COOC 2H5)2 MOL WT. 160.17 H.S. CODE TOXICITY SYNONYMS Ethyl methane dicarboxylate; Ethyl propanedioate; Diethylmalonat (German); Malonato de dietilo (Spanish); Malonate de diéthyle (French); Ethyl malonate; Malonic; Propanedioic acid diethyl ester; DERIVATION CLASSIFICATION PHYSICAL AND CHEMICAL PROPERTIES PHYSICAL STATE clear liquid MELTING POINT -50 C BOILING POINT 199 C SPECIFIC GRAVITY 1.055 SOLUBILITY IN WATER Slightly soluble pH VAPOR DENSITY 5.5 AUTOIGNITION NFPA RATINGS Health: 0; Flammability: 1; Reactivity: 0 REFRACTIVE INDEX 1.4135 FLASH POINT 93 C STABILITY Stable under ordinary conditions GENERAL DESCRIPTION & APPLICATIONS Malonic acid (also called Propanedioic Acid ) is a white crystalline C-3 dicarboxylic acid; melting at 135 -136 C; readily soluble in water, alcohol and ether; solution in water is medium-strong acidic. It can be derived by oxidizing malic acid or by the hydrolysis of cyanacetic acid. Malonic acid itself is rather unstable and has few applications. It's diethyl ester (diethyl malonate) is more important commercially. Diethyl malonate, a colourless, fragrant liquid boiling at 199 C, is prepared by the reaction of monochloroacetatic acid with methanol, carbon monoxide or by the reaction cyanoacetic acid (the half nitriled-malonic acid) with ethyl alcohol. Malonic acid and its esters contain active methylene groups which have relatively acidic alpha-protons due to H atoms adjacent to two carbonyl groups. The reactivity of its methylene group provide the sequence of reactions of alkylation, hydrolysis of the esters and decarboxylation resulting in substituted ketones. The methylene groups in 1,3-dicarboxylic acid utilize the synthesis of barbiturates; a hydrogen atom is removed by sodium ethoxide, and the derivative reacts with an alkyl halide to form a diethyl alkylmalonate. -

Microwave-Assisted Urea Catalyzed Knoevenagel Condensation of Aldehydes with = Light Brown Solid, M.P
Available online at www.banglajol.info Bangladesh J. Sci. Ind. Res. 55(2), 159-164, 2020 purification. The Chemicals 4-fluorobenzaldehyde (98%), 2-(4-nitrophenylmethylene)malononitrile (3a): yield 90%, 2-(4-Hydroxyphenylmethylene)malononitrile (3f): yield CDCl3) δ(ppm): 8.32(s, 1H), 8.01(d, J=8.4, 2H), 7.22(d, Furthermore, the treatment of aldehyde such as acknowledge the University Research Centre, SUST for Yang Y, Dong ODW, Pan W, Zhang J and Liu Q (2006), Wang C, Li Guishen S, Li G, Feng J, Li S and Xiaolu (2001), 4-hydroxybenzaldehyde (98%), 4-nitrobenzaldehyde yellowish white solid, m.p. 158-159 , (lit.160 (Sun Qi et 96%, yellow solid, m.p. 171-172 , (lit. 189-190 ( Sun J=8.4, 2H), 6.38(s, 1H, N-H), 6.11(s, 1H, N-H) . cinnamaldehyde with ethylcyanoacetate also give financial support. Facile and clean synthesis of α-alkenoyl Synthesis of 5-alkylene barbituric acid in solventless 2 Qi et al 2005 ); IR (KBr): υ (cm-1): 3354(-OH), olefinic compounds 3i under similar conditions (Table ketene-(S,S)-acetal via the aldol condensation under microwave irradiation, Chin. J. Org. Chem. (98%), 2-nitrobenzaldehyde (98%), 3-nitrobenzaldehyde al 2005)); IR (KBr): υmax: 3039(sp C-H), 2231 (C≡N), 1604 max (98%), 4-chlorobenzaldehyde (97%) and cyanoacetamide (C=C) and 1521 and 1344(N-O) cm℃-1; 1H NMR℃ (400 MHz, 3030(sp2 C-H), 2227(C≡N), and℃ 1670(C=C) cm℃-1; 1H 2-cyano-3-(4-nitrophenyl)acrylamide (3l): yield 85%, 1, Scheme 1). Both electron-rich and References reactions in water, Tetrahedron 62: 10-111. -

Ethyl Acetoacetate
21.6 The Acetoacetic Ester Synthesis Acetoacetic Ester O O C C H3C C OCH2CH3 H H Acetoacetic ester is another name for ethyl acetoacetate. The "acetoacetic ester synthesis" uses acetoacetic ester as a reactant for the preparation of ketones. Deprotonation of Ethyl Acetoacetate O O – C C + CH3CH2O H3C C OCH2CH3 H H Ethyl acetoacetate pKa ~ 11 can be converted readily to its anion with bases such as sodium ethoxide. Deprotonation of Ethyl Acetoacetate O O – C C + CH3CH2O H3C C OCH2CH3 H H Ethyl acetoacetate pKa ~ 11 can be converted readily to its anion K ~ 105 K ~ 10 with bases such as O O sodium ethoxide. C •• C + CH3CH2OH H3C –C OCH2CH3 3 2 H pKa ~ 16 Alkylation of Ethyl Acetoacetate O O The anion of ethyl C •• C acetoacetate can be H C C OCH CH 3 – C OCH2CH3 alkylated using an H alkyl halide (SN2: primary and R X secondary alkyl halides work best; tertiary alkyl halides undergo elimination). Alkylation of Ethyl Acetoacetate O O The anion of ethyl C •• C acetoacetate can be H C C OCH CH 3 – C OCH2CH3 alkylated using an H alkyl halide (SN2: primary and R X secondary alkyl O O halides work best; tertiary alkyl halides C C undergo elimination). H3C C OCH2CH3 H R Conversion to Ketone O O Saponification and C C acidification convert H3C C OH the alkylated H R derivative to the – 1. HO , H2O corresponding b-keto 2. H+ acid. O O The b-keto acid then undergoes C C decarboxylation to H C 3 C OCH2CH3 form a ketone. -
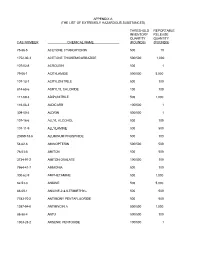
The List of Extremely Hazardous Substances)
APPENDIX A (THE LIST OF EXTREMELY HAZARDOUS SUBSTANCES) THRESHOLD REPORTABLE INVENTORY RELEASE QUANTITY QUANTITY CAS NUMBER CHEMICAL NAME (POUNDS) (POUNDS) 75-86-5 ACETONE CYANOHYDRIN 500 10 1752-30-3 ACETONE THIOSEMICARBAZIDE 500/500 1,000 107-02-8 ACROLEIN 500 1 79-06-1 ACRYLAMIDE 500/500 5,000 107-13-1 ACRYLONITRILE 500 100 814-68-6 ACRYLYL CHLORIDE 100 100 111-69-3 ADIPONITRILE 500 1,000 116-06-3 ALDICARB 100/500 1 309-00-2 ALDRIN 500/500 1 107-18-6 ALLYL ALCOHOL 500 100 107-11-9 ALLYLAMINE 500 500 20859-73-8 ALUMINUM PHOSPHIDE 500 100 54-62-6 AMINOPTERIN 500/500 500 78-53-5 AMITON 500 500 3734-97-2 AMITON OXALATE 100/500 100 7664-41-7 AMMONIA 500 100 300-62-9 AMPHETAMINE 500 1,000 62-53-3 ANILINE 500 5,000 88-05-1 ANILINE,2,4,6-TRIMETHYL- 500 500 7783-70-2 ANTIMONY PENTAFLUORIDE 500 500 1397-94-0 ANTIMYCIN A 500/500 1,000 86-88-4 ANTU 500/500 100 1303-28-2 ARSENIC PENTOXIDE 100/500 1 THRESHOLD REPORTABLE INVENTORY RELEASE QUANTITY QUANTITY CAS NUMBER CHEMICAL NAME (POUNDS) (POUNDS) 1327-53-3 ARSENOUS OXIDE 100/500 1 7784-34-1 ARSENOUS TRICHLORIDE 500 1 7784-42-1 ARSINE 100 100 2642-71-9 AZINPHOS-ETHYL 100/500 100 86-50-0 AZINPHOS-METHYL 10/500 1 98-87-3 BENZAL CHLORIDE 500 5,000 98-16-8 BENZENAMINE, 3-(TRIFLUOROMETHYL)- 500 500 100-14-1 BENZENE, 1-(CHLOROMETHYL)-4-NITRO- 500/500 500 98-05-5 BENZENEARSONIC ACID 10/500 10 3615-21-2 BENZIMIDAZOLE, 4,5-DICHLORO-2-(TRI- 500/500 500 FLUOROMETHYL)- 98-07-7 BENZOTRICHLORIDE 100 10 100-44-7 BENZYL CHLORIDE 500 100 140-29-4 BENZYL CYANIDE 500 500 15271-41-7 BICYCLO[2.2.1]HEPTANE-2-CARBONITRILE,5- -

Green Chemistry – Aspects for the Knoevenagel Reaction
2 Green Chemistry – Aspects for the Knoevenagel Reaction Ricardo Menegatti Universidade Federal de Goiás Brazil 1. Introduction Knoevenagel condensation is a classic C-C bond formation reaction in organic chemistry (Laue & Plagens, 2005). These condensations occur between aldehydes or ketones and active methylene compounds with ammonia or another amine as a catalyst in organic solvents (Knoevenagel, 1894). The Knoevenagel reaction is considered to be a modification of the aldol reaction; the main difference between these approaches is the higher acidity of the active methylene hydrogen when compared to an -carbonyl hydrogen (Smith & March, 2001). Figure 1 illustrates the condensation of a ketone (1) with a malonate compound (2) to form the Knoevenagel condensation product (3), which is then used to form the ,-unsaturated carboxylic compounds (3) and (4) (Laue & Plagens, 2005). R R R O O O O H O O O + base hydrolysis H O O O O R R (1) (2) (3) (4) Fig. 1. An example of the Knoevenagel reaction. Subsequent to the first description of the Knoevenagel reaction, changes were introduced using pyridine as the solvent and piperidine as the catalyst, which was named the Doebner Modification (Doebner, 1900). The Henry reaction is another variation of the Knoevenagel condensation that utilises compounds with an -nitro active methylene (Henry, 1895). The general mechanism for the Knoevenagel reaction, which involves deprotonation of the malonate derivative (6) by piperidine (5) and attack by the formed carbanion (8) on the carbonyl subunit (9) as an aldol reaction that forms the product (10) of the addition step is illustrated in Fig.