Polymers Functionalized with Nitrile Compounds
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

1 Abietic Acid R Abrasive Silica for Polishing DR Acenaphthene M (LC
1 abietic acid R abrasive silica for polishing DR acenaphthene M (LC) acenaphthene quinone R acenaphthylene R acetal (see 1,1-diethoxyethane) acetaldehyde M (FC) acetaldehyde-d (CH3CDO) R acetaldehyde dimethyl acetal CH acetaldoxime R acetamide M (LC) acetamidinium chloride R acetamidoacrylic acid 2- NB acetamidobenzaldehyde p- R acetamidobenzenesulfonyl chloride 4- R acetamidodeoxythioglucopyranose triacetate 2- -2- -1- -β-D- 3,4,6- AB acetamidomethylthiazole 2- -4- PB acetanilide M (LC) acetazolamide R acetdimethylamide see dimethylacetamide, N,N- acethydrazide R acetic acid M (solv) acetic anhydride M (FC) acetmethylamide see methylacetamide, N- acetoacetamide R acetoacetanilide R acetoacetic acid, lithium salt R acetobromoglucose -α-D- NB acetohydroxamic acid R acetoin R acetol (hydroxyacetone) R acetonaphthalide (α)R acetone M (solv) acetone ,A.R. M (solv) acetone-d6 RM acetone cyanohydrin R acetonedicarboxylic acid ,dimethyl ester R acetonedicarboxylic acid -1,3- R acetone dimethyl acetal see dimethoxypropane 2,2- acetonitrile M (solv) acetonitrile-d3 RM acetonylacetone see hexanedione 2,5- acetonylbenzylhydroxycoumarin (3-(α- -4- R acetophenone M (LC) acetophenone oxime R acetophenone trimethylsilyl enol ether see phenyltrimethylsilyl... acetoxyacetone (oxopropyl acetate 2-) R acetoxybenzoic acid 4- DS acetoxynaphthoic acid 6- -2- R 2 acetylacetaldehyde dimethylacetal R acetylacetone (pentanedione -2,4-) M (C) acetylbenzonitrile p- R acetylbiphenyl 4- see phenylacetophenone, p- acetyl bromide M (FC) acetylbromothiophene 2- -5- -
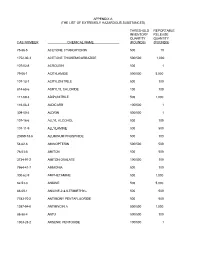
The List of Extremely Hazardous Substances)
APPENDIX A (THE LIST OF EXTREMELY HAZARDOUS SUBSTANCES) THRESHOLD REPORTABLE INVENTORY RELEASE QUANTITY QUANTITY CAS NUMBER CHEMICAL NAME (POUNDS) (POUNDS) 75-86-5 ACETONE CYANOHYDRIN 500 10 1752-30-3 ACETONE THIOSEMICARBAZIDE 500/500 1,000 107-02-8 ACROLEIN 500 1 79-06-1 ACRYLAMIDE 500/500 5,000 107-13-1 ACRYLONITRILE 500 100 814-68-6 ACRYLYL CHLORIDE 100 100 111-69-3 ADIPONITRILE 500 1,000 116-06-3 ALDICARB 100/500 1 309-00-2 ALDRIN 500/500 1 107-18-6 ALLYL ALCOHOL 500 100 107-11-9 ALLYLAMINE 500 500 20859-73-8 ALUMINUM PHOSPHIDE 500 100 54-62-6 AMINOPTERIN 500/500 500 78-53-5 AMITON 500 500 3734-97-2 AMITON OXALATE 100/500 100 7664-41-7 AMMONIA 500 100 300-62-9 AMPHETAMINE 500 1,000 62-53-3 ANILINE 500 5,000 88-05-1 ANILINE,2,4,6-TRIMETHYL- 500 500 7783-70-2 ANTIMONY PENTAFLUORIDE 500 500 1397-94-0 ANTIMYCIN A 500/500 1,000 86-88-4 ANTU 500/500 100 1303-28-2 ARSENIC PENTOXIDE 100/500 1 THRESHOLD REPORTABLE INVENTORY RELEASE QUANTITY QUANTITY CAS NUMBER CHEMICAL NAME (POUNDS) (POUNDS) 1327-53-3 ARSENOUS OXIDE 100/500 1 7784-34-1 ARSENOUS TRICHLORIDE 500 1 7784-42-1 ARSINE 100 100 2642-71-9 AZINPHOS-ETHYL 100/500 100 86-50-0 AZINPHOS-METHYL 10/500 1 98-87-3 BENZAL CHLORIDE 500 5,000 98-16-8 BENZENAMINE, 3-(TRIFLUOROMETHYL)- 500 500 100-14-1 BENZENE, 1-(CHLOROMETHYL)-4-NITRO- 500/500 500 98-05-5 BENZENEARSONIC ACID 10/500 10 3615-21-2 BENZIMIDAZOLE, 4,5-DICHLORO-2-(TRI- 500/500 500 FLUOROMETHYL)- 98-07-7 BENZOTRICHLORIDE 100 10 100-44-7 BENZYL CHLORIDE 500 100 140-29-4 BENZYL CYANIDE 500 500 15271-41-7 BICYCLO[2.2.1]HEPTANE-2-CARBONITRILE,5- -

Scientific Advisory Board
OPCW Scientific Advisory Board Eleventh Session SAB-11/1 11 – 13 February 2008 13 February 2008 Original: ENGLISH REPORT OF THE ELEVENTH SESSION OF THE SCIENTIFIC ADVISORY BOARD 1. AGENDA ITEM ONE – Opening of the Session The Scientific Advisory Board (SAB) met for its Eleventh Session from 11 to 13 February 2008 at the OPCW headquarters in The Hague, the Netherlands. The Session was opened by the Vice-Chairperson of the SAB, Mahdi Balali-Mood. The meeting was chaired by Philip Coleman of South Africa, and Mahdi Balali-Mood of the Islamic Republic of Iran served as Vice-Chairperson. A list of participants appears as Annex 1 to this report. 2. AGENDA ITEM TWO – Adoption of the agenda 2.1 The SAB adopted the following agenda for its Eleventh Session: 1. Opening of the Session 2. Adoption of the agenda 3. Tour de table to introduce new SAB Members 4. Election of the Chairperson and the Vice-Chairperson of the SAB1 5. Welcome address by the Director-General 6. Overview on developments at the OPCW since the last session of the SAB 7. Establishment of a drafting committee 8. Work of the temporary working groups: (a) Consideration of the report of the second meeting of the sampling-and-analysis temporary working group; 1 In accordance with paragraph 1.1 of the rules of procedure for the SAB and the temporary working groups of scientific experts (EC-XIII/DG.2, dated 20 October 1998) CS-2008-5438(E) distributed 28/02/2008 *CS-2008-5438.E* SAB-11/1 page 2 (b) Status report by the Industry Verification Branch on the implementation of sampling and analysis for Article VI inspections; (c) Presentation by the OPCW Laboratory; (d) Update on education and outreach; and (e) Update on the formation of the temporary working group on advances in science and technology and their potential impact on the implementation of the Convention: (i) composition of the group; and (ii) its terms of reference 9. -

House Fly Attractants and Arrestante: Screening of Chemicals Possessing Cyanide, Thiocyanate, Or Isothiocyanate Radicals
House Fly Attractants and Arrestante: Screening of Chemicals Possessing Cyanide, Thiocyanate, or Isothiocyanate Radicals Agriculture Handbook No. 403 Agricultural Research Service UNITED STATES DEPARTMENT OF AGRICULTURE Contents Page Methods 1 Results and discussion 3 Thiocyanic acid esters 8 Straight-chain nitriles 10 Propionitrile derivatives 10 Conclusions 24 Summary 25 Literature cited 26 This publication reports research involving pesticides. It does not contain recommendations for their use, nor does it imply that the uses discussed here have been registered. All uses of pesticides must be registered by appropriate State and Federal agencies before they can be recommended. CAUTION: Pesticides can be injurious to humans, domestic animals, desirable plants, and fish or other wildlife—if they are not handled or applied properly. Use all pesticides selectively and carefully. Follow recommended practices for the disposal of surplus pesticides and pesticide containers. ¿/áepé4áaUÁí^a¡eé —' ■ -"" TMK LABIL Mention of a proprietary product in this publication does not constitute a guarantee or warranty by the U.S. Department of Agriculture over other products not mentioned. Washington, D.C. Issued July 1971 For sale by the Superintendent of Documents, U.S. Government Printing Office Washington, D.C. 20402 - Price 25 cents House Fly Attractants and Arrestants: Screening of Chemicals Possessing Cyanide, Thiocyanate, or Isothiocyanate Radicals BY M. S. MAYER, Entomology Research Division, Agricultural Research Service ^ Few chemicals possessing cyanide (-CN), thio- cyanate was slightly attractive to Musca domes- eyanate (-SCN), or isothiocyanate (~NCS) radi- tica, but it was considered to be one of the better cals have been tested as attractants for the house repellents for Phormia regina (Meigen). -

IUPAC Provisional Recommendations
Preferred IUPAC Names 177 Chapter 6, Sect 66 September, 2004 P-66 Amides, imides, hydrazides, nitriles, aldehydes, their chalcogen analogues and derivatives P-66.0 Introduction P-66.1 Amides P-66.2 Imides P-66.3 Hydrazides P-66.4 Amidines, amidrazones, hydrazidines P-66.5 Nitriles P-66.6 Aldehydes P-66.0 Introduction The classes dealt with in this Section have in common the fact that their retained names are derived from those of acids by changing the ‘ic acid’ ending to a class name, for example ‘amide’, ‘ohydrazide’, ‘nitrile’ or ‘aldehyde’. Their systematic names are formed substitutively by the suffix mode using one of two types of suffix, one that includes the carbon atom, for example, ‘carbonitrile’ for −CN, and one that does not, for example, ‘-nitrile’ for −(C)N,. Amidines are named as amides, hydrazidines as hydrazides, and amidrazones as amides or hydrazides. P-66.1 Amides P-66.1.0 Introduction P-66.1.1 Primary amides P-66.1.2 Secondary and tertiary amides P-66.1.3 Chalcogen analogues of amides P-66.1.4 Lactams, lactims, sultams and sultims P-66.1.5 Amides of carbonic, oxalic, cyanic, and polycarbonic acids P-66.1.6 Polyfunctional amides P-66.1.0 Introduction Amides are derivatives of oxoacids in which each hydroxy group has been replaced by an amino or substituted amino group. Chalcogen replacement analogues are called thio-, seleno- and telluroamides. Compounds having one, two or three acyl groups on a single nitrogen atom are generically included and may be designated as primary, secondary and tertiary amides, respectively. -
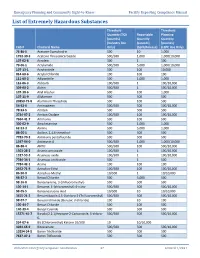
List of Extremely Hazardous Substances
Emergency Planning and Community Right-to-Know Facility Reporting Compliance Manual List of Extremely Hazardous Substances Threshold Threshold Quantity (TQ) Reportable Planning (pounds) Quantity Quantity (Industry Use (pounds) (pounds) CAS # Chemical Name Only) (Spill/Release) (LEPC Use Only) 75-86-5 Acetone Cyanohydrin 500 10 1,000 1752-30-3 Acetone Thiosemicarbazide 500/500 1,000 1,000/10,000 107-02-8 Acrolein 500 1 500 79-06-1 Acrylamide 500/500 5,000 1,000/10,000 107-13-1 Acrylonitrile 500 100 10,000 814-68-6 Acrylyl Chloride 100 100 100 111-69-3 Adiponitrile 500 1,000 1,000 116-06-3 Aldicarb 100/500 1 100/10,000 309-00-2 Aldrin 500/500 1 500/10,000 107-18-6 Allyl Alcohol 500 100 1,000 107-11-9 Allylamine 500 500 500 20859-73-8 Aluminum Phosphide 500 100 500 54-62-6 Aminopterin 500/500 500 500/10,000 78-53-5 Amiton 500 500 500 3734-97-2 Amiton Oxalate 100/500 100 100/10,000 7664-41-7 Ammonia 500 100 500 300-62-9 Amphetamine 500 1,000 1,000 62-53-3 Aniline 500 5,000 1,000 88-05-1 Aniline, 2,4,6-trimethyl- 500 500 500 7783-70-2 Antimony pentafluoride 500 500 500 1397-94-0 Antimycin A 500/500 1,000 1,000/10,000 86-88-4 ANTU 500/500 100 500/10,000 1303-28-2 Arsenic pentoxide 100/500 1 100/10,000 1327-53-3 Arsenous oxide 100/500 1 100/10,000 7784-34-1 Arsenous trichloride 500 1 500 7784-42-1 Arsine 100 100 100 2642-71-9 Azinphos-Ethyl 100/500 100 100/10,000 86-50-0 Azinphos-Methyl 10/500 1 10/10,000 98-87-3 Benzal Chloride 500 5,000 500 98-16-8 Benzenamine, 3-(trifluoromethyl)- 500 500 500 100-14-1 Benzene, 1-(chloromethyl)-4-nitro- 500/500 -

United States Patent Office 2,969,402 Patented Jan
United States Patent Office 2,969,402 Patented Jan. 24, 1961 2 preferred. In a more preferred aspect the upper tem 2,969,402 perature is about 10 C. The ratio of the three components, i.e., alkaline earth PREPARATION OF CATALYSTS FOR THE POLY metal hexammoniate, olefin oxide, and organic nitrile, MERIZATION OF EPOXDES can be varied over a wide range in the preparation of the Fred N. Hill, South Charleston, and John T. Fitzpatrick novel catalysts. The reaction is conducted, as indicated and Frederick E. Bailey, Jr., Charleston, W. Va., as previously, in an excess liquid ammonia medium. Thus, signors to Union Carbide Corporation, a corporation active catalysts can be prepared by employing from about of New York 0.3 to 1.0 mol of olefin oxide per mol of metal hexam No Drawing. Filed Dec. 29, 1958, Ser. No. 783,100 0. moniate, and from about 0.2 to 0.8 mol of organic nitrile per mol of metal hexammoniate. Extremely active 12 Claims. (CI. 260-632) catalyst can be prepared by employing from about 0.4 to 1.0 mol of olefin oxide per mol of metal hexammoniate, This invention relates to the preparation of composi and from about 0.3 to 0.6 mol of organic nitrile per mol tions which are catalytically active for the polymerization 5 of metal hexammoniate. It should be noted that the of epoxide compounds which contain a cyclic group com alkaline earth metal hexammoniate, M(NH3)6 wherein posed of two carbon atoms and one oygen atom. M can be calcium, barium, or strontium, contains alkaline Various divalent metal amides, H2N-M-NH2, and earth metal in the zero valence state. -

565.Full.Pdf
Studies on the Biological Action of Malononitriles* I. The Effect of Substituted Malononitriles on the Growth of Transplanted Tumors in Mice EMERYM. GAL,| FUNG-ÜAANFUNG,ANDDAVIDM. GREENBERG ASSISTEDBYHARRIETEZRAANDSHERBURNEF.COOK,JB. (Department of Biochemistry, School of Medicine, Unirersity of California, Berkeley, Calif.') This laboratory has been engaged for a number MATERIALS of years on a program of cancer chemotherapy re Malononitrile, by virtue of its active méthylènegroup, search guided mainly by the concept of metabolite readily condenses with carbonyl compounds to yield dinitriles. The condensation products show different degrees of saturation antagonism (9) in the selection of compounds to be and can be divided into the following groups: tested for chemotherapeutic activity. The impor 1. R-CH,CH(CN), tance attributed to the components of the nucleic 2. R-CH (CH[CN]S), acids in the genesis of neoplasms and the control of 3. R-CH:C(CN)j cellular functions has, with some measure of suc R.-. cess, oriented extensive investigations on experi 4. R2-C:C(CN)S 5. R-N:N-CH(CN)2 mental tumor chemotherapy. An outstanding ex ample is the work of Kidder and his associates (13) With a few exceptions the compounds were prepared in our on the growth-inhibiting properties of the purine laboratory according to known procedures (1, 4, 14). The car analog, 8-azaguanine. bonyl compounds used in the syntheses were, for the most part, purchased from Distillation Products Industries. The prepared Our efforts have been considerably influenced compounds were checked for purity and the elementary compo by the work of Hyden and Hartelius (12), who re sition of those which were not described in the literature deter ported that administration of malononitrile mined by micro-analysis. -

List of Lists
United States Office of Solid Waste EPA 550-B-10-001 Environmental Protection and Emergency Response May 2010 Agency www.epa.gov/emergencies LIST OF LISTS Consolidated List of Chemicals Subject to the Emergency Planning and Community Right- To-Know Act (EPCRA), Comprehensive Environmental Response, Compensation and Liability Act (CERCLA) and Section 112(r) of the Clean Air Act • EPCRA Section 302 Extremely Hazardous Substances • CERCLA Hazardous Substances • EPCRA Section 313 Toxic Chemicals • CAA 112(r) Regulated Chemicals For Accidental Release Prevention Office of Emergency Management This page intentionally left blank. TABLE OF CONTENTS Page Introduction................................................................................................................................................ i List of Lists – Conslidated List of Chemicals (by CAS #) Subject to the Emergency Planning and Community Right-to-Know Act (EPCRA), Comprehensive Environmental Response, Compensation and Liability Act (CERCLA) and Section 112(r) of the Clean Air Act ................................................. 1 Appendix A: Alphabetical Listing of Consolidated List ..................................................................... A-1 Appendix B: Radionuclides Listed Under CERCLA .......................................................................... B-1 Appendix C: RCRA Waste Streams and Unlisted Hazardous Wastes................................................ C-1 This page intentionally left blank. LIST OF LISTS Consolidated List of Chemicals -

State of Illinois Environmental Protection Agency Application for Environmental Laboratory Accreditation
State of Illinois Environmental Protection Agency Application for Environmental Laboratory Accreditation Attachment 6 Program: RCRA Field of Testing: Solid and Chemical Materials, Organic Matrix: Non Solid and Potable Chemical Accredited Water Materials Analyte: Status* Method (SW846): 1,2-Dibromo-3-chloropropane (DBCP) 8011 1,2-Dibromoethane (EDB) 8011 1,4-Dioxane 8015B 1-Butanol (n-Butyl alcohol) 8015B 1-Propanol 8015B 2-Butanone (Methyl ethyl ketone, MEK) 8015B 2-Chloroacrylonitrile 8015B 2-Methyl-1-propanol (Isobutyl alcohol) 8015B 2-Methylpyridine (2-Picoline) 8015B 2-Pentanone 8015B 2-Propanol (Isopropyl alcohol) 8015B 4-Methyl-2-pentanone (Methyl isobutyl ketone, 8015B MIBK) Acetone 8015B Acetonitrile 8015B Acrolein (Propenal) 8015B Acrylonitrile 8015B Allyl alcohol 8015B Crotonaldehyde 8015B Diesel range organics (DRO) 8015B Diethyl ether 8015B Ethanol 8015B Ethyl acetate 8015B Ethylene glycol 8015B Ethylene oxide 8015B Gasoline range organics (GRO) 8015B Hexafluoro-2-methyl-2-propanol 8015B * PI: Pending Initial Accreditation A: Accredited SP: Suspended WD: Accreditation Withdrawn Attachment 6: Page 1 of 62 Program: RCRA Field of Testing: Solid and Chemical Materials, Organic Matrix: Non Solid and Potable Chemical Accredited Water Materials Analyte: Status* Method (SW846): Hexafluoro-2-propanol 8015B Isopropyl benzene ((1-methylethyl) benzene) 8015B Methanol 8015B N-Nitrosodi-n-butylamine (N-Nitrosodibutylamine) 8015B o-Toluidine 8015B Paraldehyde 8015B Propionitrile (Ethyl cyanide) 8015B Pyridine 8015B t-Butyl alcohol 8015B -

On the Chromophore of Polyacrylonitrile. V. the Oxidation of Isobutyronitrile
72 J. BRANDRUP Macromolecules 15-20z yield in a rather impure form, bp 61' (1 mm). The gas chromatograph showed only two equally strong Peaks at 1715 and 1730 cm-l indicated ketonic impurities. peaks indicating the two isomers. The nmr spectrum agreed These could be removed partially by shaking an ethereal with the chemical structure. solution with dilute base. The remaining peak at 1730 Anal. Calcd for C6H8N2: C, 66.35; H, 7.41; N, 25.82. cm-1 could not be removed by several distillations or reac- Found: C, 66.66; H, 7.44; N, 26.07. tions with Girard's reagent. Finally, a gas chromato- graphically pufe product was obtained by chromatographing Acknowledgment. We wish to thank Mr. D. C. it on a silica gel column from a mixture of hexane and ether. Westall for experimental assistance. On the Chromophore of Polyacrylonitrile. V. The Oxidation of Isobutyronitrile J. Brandrupl Contribution No. 311 front the Chemstrand Research Center, Inc., Durham, North Carolina. Receioed October 27, 1967 ABSTRACT: The thermal degradation of polyacrylonitrile was studied with the aid of model compounds. Iso- butyronitrile was used as model for a single pendant nitrile group in polyacrylonitrile and was thermally degraded under oxygen at 150". The brown, viscous reaction product showed the presence of 30 different compounds ac- cording to gas chromatography, 16 of which have been characterized. These 16 constitute all of the major and several minor compounds. The main products were acetamide, isobutyramide, N-methylpropionamide, and the corresponding acids. In addition, a fiber-forming water-soluble polymer was recovered whose behavior and spectral characteristics agree with the structure of a polynitrone (-C=N(+O)-),. -

CSAT Top-Screen Questions OMB PRA # 1670-0007 Expires: 5/31/2011
CSAT Top-Screen Questions January 2009 Version 2.8 CSAT Top-Screen Questions OMB PRA # 1670-0007 Expires: 5/31/2011 Change Log .........................................................................................................3 CVI Authorizing Statements...............................................................................4 General ................................................................................................................6 Facility Description.................................................................................................................... 7 Facility Regulatory Mandates ................................................................................................... 7 EPA RMP Facility Identifier....................................................................................................... 9 Refinery Capacity....................................................................................................................... 9 Refinery Market Share ............................................................................................................. 10 Airport Fuels Supplier ............................................................................................................. 11 Military Installation Supplier................................................................................................... 11 Liquefied Natural Gas (LNG) Capacity................................................................................... 12 Liquefied Natural Gas Exclusion