Progress in Copper-Catalyzed Trifluoromethylation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

From Celebrex to a Novel Class of Phosphoinositide-Dependent Kinase 1 (Pdk-1) Inhibitors for Androgen-Independent Prostate Caner
FROM CELEBREX TO A NOVEL CLASS OF PHOSPHOINOSITIDE-DEPENDENT KINASE 1 (PDK-1) INHIBITORS FOR ANDROGEN-INDEPENDENT PROSTATE CANER DISSERTATION Presented in Partial Fulfillment of the requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Jiuxiang Zhu, M.S. * * * * * The Ohio State University 2005 Dissertation Committee: Approved by Professor Ching-Shih Chen, Advisor Professor Robert Brueggemeier Advisor Professor Pui-Kai (Tom) Li Graduate Program in Pharmacy Professor Matthew D. Ringel ABSTRACT Celebrex, a nonsteroidal anti-inflammatory drug (NSAID, cyclooxygenase-2 inhibitor), was reported to induce apoptosis in the prostate cancer cell line PC-3 at 50µM. Early research from our laboratory demonstrated that this apoptotic inducing effect was independent of its COX-2 inhibitory activity. Further investigation showed that PDK-1 was a major protein targeted by celebrex in PC-3 cells to induce apoptosis. However, celebrex was very weak in inhibiting PDK-1 with IC50 of 48µM. To improve its potency, two series of analogs were designed and synthesized. In the first series of 24 compounds, the 5-position methylphenyl moiety of celebrex was replaced by various aromatic ring systems to explore the optimal hydrophobic group. OSU02067 (IC50=9µM) with phenanthrene at the 5-position was the best inhibitor among this series and was selected as the lead compound for further modification. Enzyme kinetics study of PDK-1 inhibition by celebrex indicated that it competed with ATP for binding. Docking of OSU02067 into the ATP binding domain of PDK-1 showed that the sulfonamide moiety hydrogen bonded to hinge region residue Ala162. -

Shelf-Stable Electrophilic Trifluoromethylating Reagents: a Brief Historical Perspective
Shelf-stable electrophilic trifluoromethylating reagents: A brief historical perspective Norio Shibata*1, Andrej Matsnev1 and Dominique Cahard*2 Review Open Access Address: Beilstein J. Org. Chem. 2010, 6, No. 65. doi:10.3762/bjoc.6.65 1Department of Frontier Materials, Graduate School of Engineering, Nagoya Institute of Technology, Gokiso, Showa-ku, Nagoya Received: 15 March 2010 466-8555, Japan and 2UMR 6014 CNRS – C.O.B.R.A. Université et Accepted: 21 May 2010 INSA de Rouen, 1 rue Tesnière, 76130 Mont Saint Aignan, France Published: 16 June 2010 Email: Guest Editor: D. O'Hagan Norio Shibata* - [email protected]; Dominique Cahard* - [email protected] © 2010 Shibata et al; licensee Beilstein-Institut. License and terms: see end of document. * Corresponding author Keywords: asymmetric synthesis; electrophilic; fluorine; reagent; trifluoromethylation Abstract Since the discovery by Yagupolskii and co-workers that S-trifluoromethyl diarylsulfonium salts are effective for the trifluoro- methylation of thiophenolates, the design and synthesis of electrophilic trifluoromethylating reagents have been extensively researched in both academia and industry, due to the significant unique features that trifluoromethylated compounds have in phar- maceuticals, agricultural chemicals, and functional materials. Several effective reagents have been developed by the groups of Yagupolskii, Umemoto, Shreeve, Adachi, Magnier, Togni and Shibata. Due to the high stability and reactivity of these reagents, a series of Umemoto reagents, Togni reagent and Shibata reagent are now commercially available. In this review, we wish to briefly provide a historical perspective of the development of so-called “shelf-stable electrophilic trifluoromethylating reagents”, although this field is in constant development. Review The chemistry of fluoro-organic compounds is one of the areas [3]. -

AROMATIC NUCLEOPHILIC SUBSTITUTION-PART -2 Electrophilic Substitution
Dr. Tripti Gangwar AROMATIC NUCLEOPHILIC SUBSTITUTION-PART -2 Electrophilic substitution ◦ The aromatic ring acts as a nucleophile, and attacks an added electrophile E+ ◦ An electron-deficient carbocation intermediate is formed (the rate- determining step) which is then deprotonated to restore aromaticity ◦ electron-donating groups on the aromatic ring (such as -OH, -OCH3, and alkyl) make the reaction faster, since they help to stabilize the electron-poor carbocation intermediate ◦ Lewis acids can make electrophiles even more electron-poor (reactive), increasing the reaction rate. For example FeBr3 / Br2 allows bromination to occur at a useful rate on benzene, whereas Br2 by itself is slow). In fact, a substitution reaction does occur! (But, as you may suspect, this isn’t an electrophilic aromatic substitution reaction.) In this substitution reaction the C-Cl bond breaks, and a C-O bond forms on the same carbon. The species that attacks the ring is a nucleophile, not an electrophile The aromatic ring is electron-poor (electrophilic), not electron rich (nucleophilic) The “leaving group” is chlorine, not H+ The position where the nucleophile attacks is determined by where the leaving group is, not by electronic and steric factors (i.e. no mix of ortho– and para- products as with electrophilic aromatic substitution). In short, the roles of the aromatic ring and attacking species are reversed! The attacking species (CH3O–) is the nucleophile, and the ring is the electrophile. Since the nucleophile is the attacking species, this type of reaction has come to be known as nucleophilic aromatic substitution. n nucleophilic aromatic substitution (NAS), all the trends you learned in electrophilic aromatic substitution operate, but in reverse. -

The Automated Flow Synthesis of Fluorine Containing Organic Compounds
The automated flow synthesis of fluorine containing organic compounds by CHANTAL SCHOLTZ Submitted in partial fulfilment of the requirements for the degree PHILOSOPHAE DOCTOR In the Faculty of Natural & Agricultural Sciences UNIVERSITY OF PRETORIA PRETORIA Supervisor: Dr D.L. Riley February 2019 DECLARATION I, Chantal Scholtz declare that the thesis/dissertation, which I hereby submit for the degree PhD Chemistry at the University of Pretoria, is my own work and has not previously been submitted by me for a degree at this or any other tertiary institution. Signature :.......................................... Date :..................................... ii ACKNOWLEDGEMENTS I would herewith sincerely like to show my gratitude to the following individuals for their help, guidance and assistance throughout the duration of this project: My supervisor, Doctor Darren Riley, for his knowledge and commitment. Thank you for being a fantastic supervisor and allowing me the opportunity to learn so many valuable skills. My husband, Clinton, for all your love, support and patience and for always being there for me. You are the best. My family, for all the encouragement and support you gave me as well as always believing in me. I will always appreciate what you have done for me. Mr Drikus van der Westhuizen and Mr Johan Postma for their assistance at the Pelchem laboratories with product isolation and characterisation. Dr Mamoalosi Selepe for NMR spectroscopy services, Jeanette Strydom for XRF services and Gerda Ehlers at the UP library for her invaluable assistance. All my friends and colleagues for the continuous moral support, numerous helpful discussions and necessary coffee breaks. My colleagues at Chemical Process Technologies for their ongoing support and motivation, especially Dr Hannes Malan and Prof. -

S.T.E.T.Women's College, Mannargudi Semester Iii Ii M
S.T.E.T.WOMEN’S COLLEGE, MANNARGUDI SEMESTER III II M.Sc., CHEMISTRY ORGANIC CHEMISTRY - II – P16CH31 UNIT I Aliphatic nucleophilic substitution – mechanisms – SN1, SN2, SNi – ion-pair in SN1 mechanisms – neighbouring group participation, non-classical carbocations – substitutions at allylic and vinylic carbons. Reactivity – effect of structure, nucleophile, leaving group and stereochemical factors – correlation of structure with reactivity – solvent effects – rearrangements involving carbocations – Wagner-Meerwein and dienone-phenol rearrangements. Aromatic nucleophilic substitutions – SN1, SNAr, Benzyne mechanism – reactivity orientation – Ullmann, Sandmeyer and Chichibabin reaction – rearrangements involving nucleophilic substitution – Stevens – Sommelet Hauser and von-Richter rearrangements. NUCLEOPHILIC SUBSTITUTION Mechanism of Aliphatic Nucleophilic Substitution. Aliphatic nucleophilic substitution clearly involves the donation of a lone pair from the nucleophile to the tetrahedral, electrophilic carbon bonded to a halogen. For that reason, it attracts to nucleophile In organic chemistry and inorganic chemistry, nucleophilic substitution is a fundamental class of reactions in which a leaving group(nucleophile) is replaced by an electron rich compound(nucleophile). The whole molecular entity of which the electrophile and the leaving group are part is usually called the substrate. The nucleophile essentially attempts to replace the leaving group as the primary substituent in the reaction itself, as a part of another molecule. The most general form of the reaction may be given as the following: Nuc: + R-LG → R-Nuc + LG: The electron pair (:) from the nucleophile(Nuc) attacks the substrate (R-LG) forming a new 1 bond, while the leaving group (LG) departs with an electron pair. The principal product in this case is R-Nuc. The nucleophile may be electrically neutral or negatively charged, whereas the substrate is typically neutral or positively charged. -

Perfluoro-3-Ethyl-2,4-Dimethyl-3
Journal of Fluorine Chemistry 227 (2019) 109370 Contents lists available at ScienceDirect Journal of Fluorine Chemistry journal homepage: www.elsevier.com/locate/fluor Perfluoro-3-ethyl-2,4-dimethyl-3-pentyl persistent radical: A new reagent T for direct, metal-free radical trifluoromethylation and polymer initiation ⁎ ⁎ ⁎ Haibo Meia, Jianlin Hana, , Sarah Whiteb, , Greg Butlerb, Vadim A. Soloshonokc,d, a College of Chemical Engineering, Nanjing Forestry University, Nanjing, 210037, China b Oakwood Chemical, Inc. 730 Columbia Hwy. N, Estill, SC, 29918, USA c Department of Organic Chemistry I, Faculty of Chemistry, University of the Basque Country UPV/EHU, Paseo Manuel Lardizábal 3, 20018, San Sebastián, Spain d IKERBASQUE, Basque Foundation for Science, María Díaz de Haro 3, Plaza Bizkaia, 48013, Bilbao, Spain ARTICLE INFO ABSTRACT Keywords: This review comprehensively profiles perfluoro-3-ethyl-2,4-dimethyl-3-pentyl persistent radical (PPFR) asanew Fluorine reagent for radical trifluoromethylation, trifluoromethylation/fluorination and polymer initiation. The PPFRis Trifluoromethylation perfectly stable at ambient conditions, but at temperatures above 80 °C undergoes β-scission to generate tri- Persistent radical fluoromethyl radical. This property can be used to initiate various chain-polymerization or trifluoromethylation Fluorination reactions. The unique feature of this process, distinguishing it from all other known methods for ·CF3-radical Polymer generation, is that the radical is produced under neutral, inert and additive-free -

Synthesis of Organobromines As a Tool for Their Characterisation and Environmental Occurrence Assessment
Synthesis of organobromines as a tool for their characterisation and environmental occurrence assessment Andreas Rydén Department of Materials and Environmental Chemistry Stockholm University Stockholm 2013 i Doctoral Thesis 2013 Department of Materials and Environmental Chemistry Stockholm University SE-106 91 Stockholm Sweden Abstract Polybrominated diphenyl ethers (PBDEs) have been intensively used as flame retardants (FRs) and have become ubiquitous environmental pollutants. PBDEs form hydroxylated PBDEs (OH-PBDEs) as metabolites. Further, some OH-PBDEs and methoxy-PBDEs (MeO-PBDEs) are natural products. These are all compounds of environmental and health concern and it is therefore important to confirm their identity and to assess their environmental levels and toxicities. Hence, it is vital to obtain authentic reference standards of individual PBDEs and OH/MeO-PBDEs. The thesis main aim was to develop synthesis methods of congener specific PBDEs, OH- and MeO-PBDEs. The second aim was to identify and quantify PBDEs, OH- and MeO-PBDEs in environmental samples. The third was to propose an abbreviation system for FRs. O-Arylation of brominated phenols, using either symmetrical or unsymmetrical brominated diphenyliodonium salts, was selected for synthesis of PBDEs and OH- /MeO-PBDEs. A total of 16 MeO-PBDEs, 11 OH-PBDEs, 1 diMeO-PBDE and 1 EtO-MeO-PBDE were synthesised. Three novel unsymmetrical diaryliodonium triflates were synthesised and used in synthesis. Optimisations were made to construct a reliable general method for congener specific PBDE synthesis, which was used in the synthesis of 8 representative PBDE congeners. The products were generally characterised by electron ionisation mass spectrometry (EIMS) and nuclear magnetic resonance (NMR) spectroscopy. -

Advances in Catalytic Enantioselective Fluorination, Mono‑,Di‑, and Trifluoromethylation, and Trifluoromethylthiolation Reactions ‡ ‡ † Xiaoyu Yang, Tao Wu, Robert J
Review pubs.acs.org/CR Advances in Catalytic Enantioselective Fluorination, Mono‑,Di‑, and Trifluoromethylation, and Trifluoromethylthiolation Reactions ‡ ‡ † Xiaoyu Yang, Tao Wu, Robert J. Phipps, and F. Dean Toste* Department of Chemistry, University of California, Berkeley, California 94720, United States 6. Catalytic Enantioselective Trifluoromethylthiola- tion 865 7. Summary and Outlook 866 Author Information 866 Corresponding Author 866 Present Address 866 Author Contributions 866 Notes 866 CONTENTS Biographies 866 Acknowledgments 867 1. Introduction 826 References 867 2. Catalytic Enantioselective Fluorination 827 2.1. Electrophilic Fluorination 827 2.1.1. Metal-Catalyzed Fluorination Involving 1. INTRODUCTION Enolates 827 Despite being largely absent from natural products and 2.1.2. Metal-Catalyzed Fluorination Not In- biological processes, fluorine plays a conspicuous and volving Enolates 834 increasingly important role within pharmaceuticals and agro- 2.1.3. Organocatalytic Electrophilic Fluorina- − chemicals, as well as in materials science.1a c Indeed, as many as tion 835 35% of agrochemicals and 20% of pharmaceuticals on the 2.1.4. Fluorination Using Multiple Catalysts 846 market contain fluorine.1d Fluorine is the most electronegative 2.1.5. One-Pot and Tandem Processes 848 element in the periodic table, and the introduction of one or 2.2. Nucleophilic Fluorination 850 more fluorine atoms into a molecule can result in greatly 3. Catalytic Enantioselective Trifluoromethylation perturbed properties. Fluorine substituents can potentially and Perfluoroalkylation 853 impact a number of variables, such as the acidity or basicity of 3.1. Asymmetric Nucleophilic Trifluoromethyla- neighboring groups, dipole moment, and properties such as tion 853 lipophilicity, metabolic stability, and bioavailability. The 3.1.1. Overview of Nucleophilic Trifluorome- multitude of effects that can arise from the introduction of thylation 853 fluorine in small molecules in the context of medicinal 3.1.2. -

CV-PSB-August 2020
Curriculum Vitae Phil S. Baran Appointment: Scripps Research Professor, Department of Chemistry 10550 North Torrey Pines Road, BCC-436 La Jolla, California 92037 Telephone: (858) 784-7373 Facsimile: (858) 784-7575 Email: [email protected] Website: www.scripps.edu/chem/baran/ January, 2013 Darlene Shiley Professor of Chemistry April, 2009 Member, Skaggs Institute for Chemical Biology June, 2008 Professor of Chemistry July, 2006 Associate Professor of Chemistry (with Tenure) June, 2003 Assistant Professor of Chemistry Date/Place of Birth: 10 Aug 1977 / Denville, NJ, USA Citizenship: United States Education 2001 – 2003 Postdoctoral Associate Advisor: Professor E.J. Corey Harvard University, Cambridge, Massachusetts 1997 – 2001 Ph.D. Graduate Student in Chemistry Advisor: Professor K.C. Nicolaou The Scripps Research Institute, La Jolla, California 1995 – 1997 B.S. with Honors in Chemistry Advisor: Professor D.I. Schuster New York University, New York, New York 1991 – 1995 Simultaneous high school graduation from Mt. Dora High School and A.A. degree with honors, Lake Sumter Community College, Florida Awards • Inhoffen Medal, 2019 • Manchot Research Professorship, 2017 • Member, The National Academy of Sciences, 2017 • Emanuel Merck Lectureship, 2017 • Blavatnik National Laureate in Chemistry, 2016 • ACS Elias J. Corey Award, 2016 • Member, American Academy of Arts and Sciences, 2015 • College of Arts and Science Alumni Distinguished Service Award, New York University, 2015 • Reagent of the Year Award (EROS), 2015 • Mukaiyama Award, 2014 • -
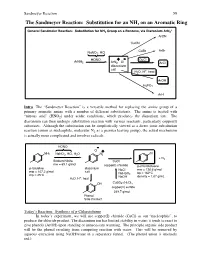
The Sandmeyer Reaction: Substitution for an NH2 on an Aromatic Ring
Sandmeyer Reaction 59 The Sandmeyer Reaction: Substitution for an NH2 on an Aromatic Ring + General Sandmeier Reaction: Substitution for NH2 Group on a Benzene, via Diazonium ArN2 ArCN CuCN CuBr ArBr NaNO2, HCl HONO + - CuCl ArNH2 ArN2 Cl ArCl diazonium salt + H2O, H , heat ArOH H3PO2 ArH Intro The “Sandmeyer Reaction” is a versatile method for replacing the amine group of a primary aromatic amine with a number of different substitutents. The amine is treated with “nitrous acid” (HNO2) under acidic conditions, which produces the diazonium ion. The diazonium can then undergo substitution reaction with various reactants, particularly copper(I) substrates. Although the substitution can be simplistically viewed as a direct ionic substitution reaction (anion as nucleophile, molecular N2 as a premier leaving group), the actual mechanism is actually more complicated and involves radicals. HONO - Cl + NH2 NaNO2, HCl, H2O N2 Cl + N2 Sodium Nitrite CuCl mw = 69.1 g/mol copper(I) chloride p-chlorotoluene p-toluidine diazonium NaCl mw = 126.6 g/mol mw = 107.2 g/mol salt NaHSO bp = 162ºC mp = 45ºC 3 + NaOH density = 1.07 g/mL H2O, H , heat OH CuSO4-(H2O)5 copper(II) sulfate 249.7 g/mol Phenol Side Product Today’s Reaction: Synthesis of p-Chlorotoluene In today’s experiment, we will use copper(I) chloride (CuCl) as our “nucleophile”, to produce the chloride product. The diazonium ion has limited stability in water; it tends to react to give phenols (ArOH) upon standing or unnecessary warming. The principle organic side product will be the phenol resulting from competing reaction with water. -

Development of Palladium-Catalyzed Decarboxylative Allylation of Electron-Deficient Sulfones: Method Development and Mechanistic Studies
Development of Palladium-Catalyzed Decarboxylative Allylation of Electron-Deficient Sulfones: Method Development and Mechanistic Studies by Monica Anne Gill A thesis submitted to the Faculty of Graduate and Postdoctoral Affairs in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Chemistry Carleton University Ottawa, Ontario © 2015, Monica Anne Gill Abstract Palladium-catalyzed decarboxylative allylation is a powerful method of carbon-carbon bond construction. This methodology relies on an electron- withdrawing group to promote the reaction. The use of sulfones in decarboxylative allylation has been explored using the trifluoromethylsulfonyl (triflyl) group, as well as the bis(3,5-trifluoromethyl)phenylsulfonyl (BTMP) group. These substrates are highly reactive at room temperature (triflyl) and 50 oC (BTMP sulfones). A detailed mechanistic study using deuterium-labelled substrates was performed to understand the origin of the protonation side-product. It was proposed that a β- hydride elimination from the η1 allyl on palladium could generate a palladium hydride intermediate along with an allene. Although small amounts of deuterium incorporation were observed in the protonated products, the proposed mechanism could not be the major pathway. Using isotopically labelled ligand, however, all protonation was supressed. This suggests that the origin of the proton is actually from the ligand and that kinetic isotope effects may be responsible for inhibiting the protonation pathway with labelled ligand. ii Acknowledgments I would like to thank Dr. Jeff Manthorpe for giving me the chance to pursue my PhD in his lab. I always appreciated Jeff’s enthusiasm for organic chemistry as well as his vast knowledge of the subject. -

Sandmeyer Reaction
Sandmeyer reaction The Sandmeyer reaction is a chemical reaction used to synthesize aryl halides from Sandmeyer reaction aryl diazonium salts using copper salts as reagents or catalysts.[1][2][3] It is an example Named after Traugott of a radical-nucleophilic aromatic substitution. The Sandmeyer reaction provides a method through which one can perform unique transformations on benzene, such as Sandmeyer halogenation, cyanation, trifluoromethylation, and hydroxylation. Reaction type Substitution reaction Identifiers Organic sandmeyer- Chemistry reaction The reaction was discovered in 1884 by Swiss chemist Traugott Sandmeyer, when he Portal attempted to synthesize phenylacetylene from benzenediazonium chloride and cuprous RSC ontology RXNO:0000021 [4] acetylide. Instead, the main product he isolated was phenyl chloride. In modern ID times, the Sandmeyer reaction refers to any method for substitution of an aromatic amino group via preparation of its diazonium salt followed by its displacement with a nucleophile in the presence of catalytic copper(I) salts. (Due to the low cost of copper salts, a stoichiometric amount is often employed for better reactivity even when catalysis is possible.) The most commonly employed Sandmeyer reactions are the chlorination, bromination, cyanation, and hydroxylation reactions using CuCl, CuBr, CuCN, and Cu2O, respectively. More recently, trifluoromethylation of diazonium salts has been developed and is referred to as a 'Sandmeyer-type' reaction. Diazonium salts also react with boronates, iodide, thiols, water,