'Identification of Synthetic Lethal Gene Interactions with REV3 in Lung
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Lorena Novoa-Aponte
A Strain: fl/fl ∆hep Strain: fl/fl ∆hep AnalysisAAV: ofLuc the ironLuc chaperonePCBP1-WT PCBP1PCBP1-∆Fe interactome:PCBP1-∆RNA PCBP1 variant intersection of DNA repair and iron traffickingAAV: Luc Luc WT ∆Fe ∆RNA PCBP1 −40 Lorena Novoa-Aponte a*, Sarju J. Patel a, Olga Protchenko a, James Wohlschlegel b, Caroline C. Philpott a −40 PCBP1, IHC PCBP1, GAPDH - a Genetics and Metabolism Section,a NIDDK, NIH, Bethesda, MD, USA. b Department of Biological Chemistry, UCLA, Los Angeles, CA, USA C.C. Philpott, et al. *[email protected] 80 B P = 0.0262 60 40 1. Iron is essential but toxic 2. Increased DNA damage in cells lacking PCBP1 ns H&E ? + 20 C.C. Philpott, et al. Iron is used as an essential cofactor by several enzymes involved in DNA ⦿ Increased TUNEL in cells and tissue livers from mice lacking PCBP1. ? replication and repair. However, unchaperoned iron promotes redox ⦿ PCBP1 binds both, iron and single-stranded nucleicTG, nmol/mg protein 0 acids. stress that may affect DNA stability. Strain: Δhep Δhep Δhep ? ⦿ The iron binding activity of PCBP1 controls suppressionAAV: of DNA damage C WT ∆Fe ∆RNA PCBP1 variant ? Iron storage HEK293 cells Mouse Liver ? A 8 siNT+P1var 100 ? A 8 var siPCBP1 2 ? Mammals use the iron siNT+P1 ns var Fe-S cluster assembly 6 siPCBP1+P1siPCBP1 80 ADI1 TUNEL chaperone PCBP1 to var ? 6 siPCBP1+P1 metalate several iron- 60 = 0.0468 = 0.0354 = 0.0465 cell / mm population P P P population + dependent enzymes + 4 + 4 ? 40 Degradation of HIF1α Fig. 3. Iron chaperone-mediated handling of cytosolic labile iron pool. -

Whole Egg Consumption Increases Gene Expression Within the Glutathione Pathway in the Liver of Zucker Diabetic Fatty Rats
Food Science and Human Nutrition Publications Food Science and Human Nutrition 11-3-2020 Whole egg consumption increases gene expression within the glutathione pathway in the liver of Zucker Diabetic Fatty rats Joe L. Webb Iowa State University Amanda E. Bries Iowa State University, [email protected] Brooke Vogel Iowa State University Claudia Carrillo Iowa State University, [email protected] Lily Harvison Iowa State University, [email protected] See next page for additional authors Follow this and additional works at: https://lib.dr.iastate.edu/fshn_hs_pubs Part of the Dietetics and Clinical Nutrition Commons, Endocrinology, Diabetes, and Metabolism Commons, Exercise Science Commons, Food Chemistry Commons, Human and Clinical Nutrition Commons, and the Molecular, Genetic, and Biochemical Nutrition Commons The complete bibliographic information for this item can be found at https://lib.dr.iastate.edu/ fshn_hs_pubs/38. For information on how to cite this item, please visit http://lib.dr.iastate.edu/ howtocite.html. This Article is brought to you for free and open access by the Food Science and Human Nutrition at Iowa State University Digital Repository. It has been accepted for inclusion in Food Science and Human Nutrition Publications by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. Whole egg consumption increases gene expression within the glutathione pathway in the liver of Zucker Diabetic Fatty rats Abstract Nutrigenomic evidence supports the idea that Type 2 Diabetes Mellitus (T2DM) arises due to the interactions between the transcriptome, individual genetic profiles, lifestyle, and diet. Since eggs are a nutrient dense food containing bioactive ingredients that modify gene expression, our goal was to examine the role of whole egg consumption on the transcriptome during T2DM. -

Qt38n028mr Nosplash A3e1d84
! ""! ACKNOWLEDGEMENTS I dedicate this thesis to my parents who inspired me to become a scientist through invigorating scientific discussions at the dinner table even when I was too young to understand what the hippocampus was. They also prepared me for the ups and downs of science and supported me through all of these experiences. I would like to thank my advisor Dr. Elizabeth Blackburn and my thesis committee members Dr. Eric Verdin, and Dr. Emmanuelle Passegue. Liz created a nurturing and supportive environment for me to explore my own ideas, while at the same time teaching me how to love science, test my questions, and of course provide endless ways to think about telomeres and telomerase. Eric and Emmanuelle both gave specific critical advice about the proper experiments for T cells and both volunteered their lab members for further critical advice. I always felt inspired with a sense of direction after thesis committee meetings. The Blackburn lab is full of smart and dedicated scientists whom I am thankful for their support. Specifically Dr. Shang Li and Dr. Brad Stohr for their stimulating scientific debates and “arguments.” Dr. Jue Lin, Dana Smith, Kyle Lapham, Dr. Tet Matsuguchi, and Kyle Jay for their friendships and discussions about what my data could possibly mean. Dr. Eva Samal for teaching me molecular biology techniques and putting up with my late night lab exercises. Beth Cimini for her expertise with microscopy, FACs, singing, and most of all for being a caring and supportive friend. Finally, I would like to thank Dr. Imke Listerman, my scientific partner for most of the breast cancer experiments. -

The Inverse Agonist DG172 Triggers a Pparβ/Δ-Independent Myeloid Lineage Shift and Promotes GM-CSF/IL-4-Induced Dendritic Cell Differentiation
Downloaded from molpharm.aspetjournals.org at ASPET Journals on September 26, 2021 1 MOL #94672 #94672 MOL -independent myeloid lineage shift and -independent myeloid lineage δ / β nagel, Wolfgang Meissner, Gavin Giehl, Cornelia Giehl, Cornelia Gavin Wolfgang Meissner, nagel, This article has not been copyedited and formatted. The final version may differ from this version. This article has not been copyedited and formatted. The final version may differ from this version. This article has not been copyedited and formatted. The final version may differ from this version. This article has not been copyedited and formatted. The final version may differ from this version. This article has not been copyedited and formatted. The final version may differ from this version. This article has not been copyedited and formatted. The final version may differ from this version. This article has not been copyedited and formatted. The final version may differ from this version. This article has not been copyedited and formatted. The final version may differ from this version. This article has not been copyedited and formatted. The final version may differ from this version. This article has not been copyedited and formatted. The final version may differ from this version. This article has not been copyedited and formatted. The final version may differ from this version. This article has not been copyedited and formatted. The final version may differ from this version. This article has not been copyedited and formatted. The final version may differ from this version. This article has not been copyedited and formatted. The final version may differ from this version. -

Hypermutation of DPYD Deregulates Pyrimidine Metabolism and Promotes Malignant Progression Lauren Edwards, Rohit Gupta, and Fabian Volker Filipp
Published OnlineFirst November 25, 2015; DOI: 10.1158/1541-7786.MCR-15-0403 Genomics Molecular Cancer Research Hypermutation of DPYD Deregulates Pyrimidine Metabolism and Promotes Malignant Progression Lauren Edwards, Rohit Gupta, and Fabian Volker Filipp Abstract New strategies are needed to diagnose and target human the background mutation rate. Structural analysis of the DPYD melanoma. To this end, genomic analyses was performed to protein dimer reveals a potential hotspot of recurring somatic assess somatic mutations and gene expression signatures using mutations in the ligand-binding sites as well as the interfaces of a large cohort of human skin cutaneous melanoma (SKCM) protein domains that mediated electron transfer. Somatic muta- patients from The Cancer Genome Atlas (TCGA) project to tions of DPYD are associated with upregulation of pyrimidine identify critical differences between primary and metastatic degradation, nucleotide synthesis, and nucleic acid processing tumors. Interestingly, pyrimidine metabolism is one of the major while salvage and nucleotide conversion is downregulated in pathways to be significantly enriched and deregulated at the TCGA SKCM. transcriptional level in melanoma progression. In addition, dihy- dropyrimidine dehydrogenase (DPYD) and other important Implications: At a systems biology level, somatic mutations of pyrimidine-related genes: DPYS, AK9, CAD, CANT1, ENTPD1, DPYD cause a switch in pyrimidine metabolism and promote NME6, NT5C1A, POLE, POLQ, POLR3B, PRIM2, REV3L, and gene expression of pyrimidine enzymes toward malignant pro- UPP2 are significantly enriched in somatic mutations relative to gression. Mol Cancer Res; 14(2); 196–206. Ó2015 AACR. Introduction Pyrimidine synthesis is a key metabolic bottleneck important for DNA replication in tumor cells and, therefore, represents a Cancer cells take advantage of distinct metabolic pathways valuable diagnostic and therapeutic target. -

Investigating the Impact of Telomere Dysfunction on the Chronic Lymphocytic Leukaemia Genome
INVESTIGATING THE IMPACT OF TELOMERE DYSFUNCTION ON THE CHRONIC LYMPHOCYTIC LEUKAEMIA GENOME Laura Escudero-Monreal A thesis submitted for the degree of Doctor of Philosophy September 2017 Institute of Cancer and Genetics School of Medicine Cardiff University Funded by Cancer Research Wales and Cardiff University I dedicate this Ph.D. thesis to everyone who suffers from cancer, for I hope one day, science can fight and win this battle. Laura Escudero-Monreal NOTICE OF SUBMISSION DECLARATION This work has not been submitted in substance for any other degree or award at this or any other university or place of learning, nor is being submitted concurrently in candidature for any degree or other award. Signed: Date: STATEMENT 1 This thesis is being submitted in partial fulfilment of the requirements for the degree of PhD. Signed: Date: STATEMENT 2 This thesis is the result of my own independent work/investigation, except where otherwise stated, and the thesis has not been edited by a third party beyond what is permitted by Cardiff University’s Policy on the Use of Third Party Editors by Research Degree Students. Other sources are acknowledged by explicit references. The views expressed are my own. Signed: Date: STATEMENT 3 I hereby give consent for my thesis, if accepted, to be available online in the University’s Open Access repository and for inter-library loan, and for the title and summary to be made available to outside organisations. Signed: Date: STATEMENT 4: PREVIOUSLY APPROVED BAR ON ACCESS I hereby give consent for my thesis, if accepted, to be available online in the University’s Open Access repository and for inter-library loans after expiry of a bar on access previously approved by the Academic Standards & Quality Committee. -

Thesis Template
Characterisation of the Co-chaperone Small Glutamine-rich Tetratricopeptide Repeat containing protein alpha as a Regulator of Androgen Receptor Activity in Prostate Cancer Cells A thesis submitted to the University of Adelaide in total fulfilment of the requirements for the degree of Doctor of Philosophy by ANDREW PAUL TROTTA B.Sc. (Mol. Biol.), B.Sc. (Hons) Department of Medicine The University of Adelaide Adelaide, South Australia July 2011 This thesis is dedicated to my mum and dad. Thank you for all your love and support. DECLARATION I ACKNOWLEDGEMENTS II ABBREVIATIONS V ABSTRACT X CHAPTER 1: INTRODUCTION 2 1.1 Overview 2 1.2 Development of the prostate 4 1.2.1 Androgen physiology 4 1.2.2 Development of the normal prostate 5 1.3 Prostate cancer and progression 9 1.3.1 Pathogenesis 9 1.4 Diagnosis 10 1.4.1 Clinically localized and advanced disease 11 1.5 Treatment 12 1.5.1 Localised Disease 12 1.5.2 Metastatic Disease 12 1.6 The androgen signalling axis 14 1.6.1 The androgen receptor 15 1.6.2 The androgen receptor gene 16 1.6.3 The androgen receptor protein and domains 19 1.7 Androgen receptor co-regulators 24 1.7.1 Co-activators 24 1.7.2 Co-repressors 25 1.7.3 Chaperones 26 1.8 The molecular chaperone complex and androgen receptor maturation 27 1.8.1 Chaperones involved in ligand binding and nuclear translocation 32 1.8.2 Chaperones and transcriptional activation 37 1.9 Chaperones in prostate cancer 37 1.10 Chaperones as therapeutic targets 39 1.11 Tetratricopeptide repeat containing co-chaperones 40 1.11.1 Structure of TPR domain -

Genome-Wide Analysis of Organ-Preferential Metastasis of Human Small Cell Lung Cancer in Mice
Vol. 1, 485–499, May 2003 Molecular Cancer Research 485 Genome-Wide Analysis of Organ-Preferential Metastasis of Human Small Cell Lung Cancer in Mice Soji Kakiuchi,1 Yataro Daigo,1 Tatsuhiko Tsunoda,2 Seiji Yano,3 Saburo Sone,3 and Yusuke Nakamura1 1Laboratory of Molecular Medicine, Human Genome Center, Institute of Medical Science, The University of Tokyo, Tokyo, Japan; 2Laboratory for Medical Informatics, SNP Research Center, Riken (Institute of Physical and Chemical Research), Tokyo, Japan; and 3Department of Internal Medicine and Molecular Therapeutics, The University of Tokushima School of Medicine, Tokushima, Japan Abstract Molecular interactions between cancer cells and their Although a number of molecules have been implicated in microenvironment(s) play important roles throughout the the process of cancer metastasis, the organ-selective multiple steps of metastasis (5). Blood flow and other nature of cancer cells is still poorly understood. To environmental factors influence the dissemination of cancer investigate this issue, we established a metastasis model cells to specific organs (6). However, the organ specificity of in mice with multiple organ dissemination by i.v. injection metastasis (i.e., some organs preferentially permit migration, of human small cell lung cancer (SBC-5) cells. We invasion, and growth of specific cancer cells, but others do not) analyzed gene-expression profiles of 25 metastatic is a crucial determinant of metastatic outcome, and proteins lesions from four organs (lung, liver, kidney, and bone) involved in the metastatic process are considered to be using a cDNA microarray representing 23,040 genes and promising therapeutic targets. extracted 435 genes that seemed to reflect the organ More than a century ago, Stephen Paget suggested that the specificity of the metastatic cells and the cross-talk distribution of metastases was not determined by chance, but between cancer cells and microenvironment. -

Roles of Human POLD1 and POLD3 in Genome Stability Emanuela Tumini, Sonia Barroso, Carmen Pérez-Calero & Andrés Aguilera
www.nature.com/scientificreports OPEN Roles of human POLD1 and POLD3 in genome stability Emanuela Tumini, Sonia Barroso, Carmen Pérez-Calero & Andrés Aguilera DNA replication is essential for cellular proliferation. If improperly controlled it can constitute a major Received: 11 July 2016 source of genome instability, frequently associated with cancer and aging. POLD1 is the catalytic Accepted: 16 November 2016 subunit and POLD3 is an accessory subunit of the replicative Pol δ polymerase, which also functions in Published: 15 December 2016 DNA repair, as well as the translesion synthesis polymerase Pol ζ, whose catalytic subunit is REV3L. In cells depleted of POLD1 or POLD3 we found a differential but general increase in genome instability as manifested by DNA breaks, S-phase progression impairment and chromosome abnormalities. Importantly, we showed that both proteins are needed to maintain the proper amount of active replication origins and that POLD3-depletion causes anaphase bridges accumulation. In addition, POLD3-associated DNA damage showed to be dependent on RNA-DNA hybrids pointing toward an additional and specific role of this subunit in genome stability. Interestingly, a similar increase in RNA-DNA hybrids-dependent genome instability was observed in REV3L-depleted cells. Our findings demonstrate a key role of POLD1 and POLD3 in genome stability and S-phase progression revealing RNA-DNA hybrids-dependent effects for POLD3 that might be partly due to its Polζ interaction. DNA replication is an essential process in which DNA is duplicated and passed on to daughter cells, allowing the transmission of genetic information. To safeguard its integrity, cells have developed sophisticated mechanisms that constitute the DNA damage response (DDR) pathway. -

Transforming Growth Factor ß1-Mediated Functional Inhibition Of
Myelodysplastic Syndromes SUPPLEMENTARY APPENDIX Transforming growth factor 1- mediated functional inhibition of mesenchymal stromal celβls in myelodysplastic syndromes and acute myeloid leukemia Stefanie Geyh, 1* Manuel Rodríguez-Paredes, 1,2 * Paul Jäger, 1 Annemarie Koch, 1 Felix Bormann, 2 Julian Gutekunst, 2 Christoph Zilkens, 3 Ulrich Germing, 1 Guido Kobbe, 1 Frank Lyko, 2 Rainer Haas 1 and Thomas Schroeder 1 1Department of Hematology, Oncology and Clinical Immunology, University of Duesseldorf, Medical Faculty; 2Division of Epigenetics, DKFZ- ZMBH Alliance, German Cancer Research Center, Heidelberg and 3Department of Orthopedic Surgery, University of Duesseldorf, Medical Faculty, Germany *SG and MR-P contributed equally to this work. ©2018 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol. 2017.186734 Received: December 19, 2017. Accepted: May 14, 2018. Pre-published: May 17, 2018. Correspondence: [email protected] Figure S1 Downregulated genes Downregulated genes Upregulated Figure S1. Heatmaps showing the 50 most upregulated and downregulated genes between the 3 healthy MSC controls and the 9 RCMD-, RAEB- and AML-derived MSC samples. Color scale depicts the rlog-transformed FPKM values for each gene and every sample. Figure S2 Downregulated genes Downregulated genes Upregulated Figure S2. Heatmaps showing the 50 most upregulated and downregulated genes between the 3 healthy MSC controls and the 3 RCMD, RAEB and AML MSC samples, respectively. Color scales depict the rlog-transformed FPKM values for each gene and every sample. Figure S3 A. B. 0.0015 *** ** <-3 -2 0.0010 RCMD RAEB AML -1 0 1 0.0005 Log2FC LTF 2 CCL26/GAPDH INHBB >3 0.0000 TGFB2 y S h D ML M A ealt ll LTF H a EGF 0.003 *** ** INHBB TGFB2 0.002 INHBB IGFBP7 0.001 GDF11 LIF/GAPDH BMP1 0.000 y L th M TNFSF12 l A FGF13 ea ll MDS H a FGF13 0.0015 * TNFSF10 TNFSF10 0.0010 0.0005 SPP1/GAPDH 0.0000 y th l AML ea H all MDS Figure S3. -
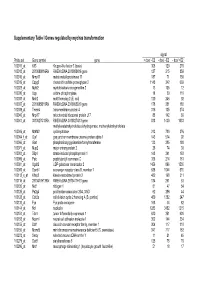
Supplementary Table I Genes Regulated by Myc/Ras Transformation
Supplementary Table I Genes regulated by myc/ras transformation signal Probe set Gene symbol gene + dox/ - E2 - dox/ - E2 - dox/ +E2 100011_at Klf3 Kruppel-like factor 3 (basic) 308 120 270 100013_at 2010008K16Rik RIKEN cDNA 2010008K16 gene 127 215 859 100016_at Mmp11 matrix metalloproteinase 11 187 71 150 100019_at Cspg2 chondroitin sulfate proteoglycan 2 1148 342 669 100023_at Mybl2 myeloblastosis oncogene-like 2 13 105 12 100030_at Upp uridine phosphorylase 18 39 110 100033_at Msh2 mutS homolog 2 (E. coli) 120 246 92 100037_at 2310005B10Rik RIKEN cDNA 2310005B10 gene 178 351 188 100039_at Tmem4 transmembrane protein 4 316 135 274 100040_at Mrpl17 mitochondrial ribosomal protein L17 88 142 89 100041_at 3010027G13Rik RIKEN cDNA 3010027G13 gene 818 1430 1033 methylenetetrahydrofolate dehydrogenase, methenyltetrahydrofolate 100046_at Mthfd2 cyclohydrolase 213 720 376 100064_f_at Gja1 gap junction membrane channel protein alpha 1 143 574 92 100066_at Gart phosphoribosylglycinamide formyltransferase 133 385 180 100071_at Mup2 major urinary protein 2 26 74 30 100081_at Stip1 stress-induced phosphoprotein 1 148 341 163 100089_at Ppic peptidylprolyl isomerase C 358 214 181 100091_at Ugalt2 UDP-galactose translocator 2 1456 896 1530 100095_at Scarb1 scavenger receptor class B, member 1 638 1044 570 100113_s_at Kifap3 kinesin-associated protein 3 482 168 311 100116_at 2810417H13Rik RIKEN cDNA 2810417H13 gene 134 261 53 100120_at Nid1 nidogen 1 81 47 54 100125_at Pa2g4 proliferation-associated 2G4, 38kD 62 286 44 100128_at Cdc2a cell division cycle 2 homolog A (S. pombe) 459 1382 247 100133_at Fyn Fyn proto-oncogene 168 44 82 100144_at Ncl nucleolin 1283 3452 1215 100151_at Tde1 tumor differentially expressed 1 620 351 620 100153_at Ncam1 neural cell adhesion molecule 1 302 144 234 100155_at Ddr1 discoidin domain receptor family, member 1 304 117 310 100156_at Mcmd5 mini chromosome maintenance deficient 5 (S.