Towards Reconstitution of Human Rnase P Protein Subunits with Human and Bacterial Rnase P Rnas
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Mouse Pop1 Conditional Knockout Project (CRISPR/Cas9)
https://www.alphaknockout.com Mouse Pop1 Conditional Knockout Project (CRISPR/Cas9) Objective: To create a Pop1 conditional knockout Mouse model (C57BL/6J) by CRISPR/Cas-mediated genome engineering. Strategy summary: The Pop1 gene (NCBI Reference Sequence: NM_152894 ; Ensembl: ENSMUSG00000022325 ) is located on Mouse chromosome 15. 17 exons are identified, with the ATG start codon in exon 2 and the TGA stop codon in exon 17 (Transcript: ENSMUST00000052290). Exon 4~5 will be selected as conditional knockout region (cKO region). Deletion of this region should result in the loss of function of the Mouse Pop1 gene. To engineer the targeting vector, homologous arms and cKO region will be generated by PCR using BAC clone RP23-365B13 as template. Cas9, gRNA and targeting vector will be co-injected into fertilized eggs for cKO Mouse production. The pups will be genotyped by PCR followed by sequencing analysis. Note: Exon 4 starts from about 9.92% of the coding region. The knockout of Exon 4~5 will result in frameshift of the gene. The size of intron 3 for 5'-loxP site insertion: 2480 bp, and the size of intron 5 for 3'-loxP site insertion: 1452 bp. The size of effective cKO region: ~2209 bp. The cKO region does not have any other known gene. Page 1 of 8 https://www.alphaknockout.com Overview of the Targeting Strategy Wildtype allele 5' gRNA region gRNA region 3' 1 4 5 6 7 17 Targeting vector Targeted allele Constitutive KO allele (After Cre recombination) Legends Exon of mouse Pop1 Homology arm cKO region loxP site Page 2 of 8 https://www.alphaknockout.com Overview of the Dot Plot Window size: 10 bp Forward Reverse Complement Sequence 12 Note: The sequence of homologous arms and cKO region is aligned with itself to determine if there are tandem repeats. -

Rpp2, an Essential Protein Subunit of Nuclear Rnase P, Is Required for Processing of Precursor Trnas and 35S Precursor Rrna in Saccharomyces Cerevisiae
Proc. Natl. Acad. Sci. USA Vol. 95, pp. 6716–6721, June 1998 Biochemistry Rpp2, an essential protein subunit of nuclear RNase P, is required for processing of precursor tRNAs and 35S precursor rRNA in Saccharomyces cerevisiae VIKTOR STOLC*, ALEXANDER KATZ†, AND SIDNEY ALTMAN†‡ †Department of Biology, Yale University, New Haven, CT 06520; and *Department of Cell Biology, Yale University School of Medicine, New Haven, CT 06510 Contributed by Sidney Altman, March 31, 1998 ABSTRACT RPP2, an essential gene that encodes a 15.8- has been suggested that RNase P is an ancestor of RNase kDa protein subunit of nuclear RNase P, has been identified MRP, because RNase MRP has been found only in eukaryotes in the genome of Saccharomyces cerevisiae. Rpp2 was detected (20). Yeast RNase MRP functions in processing of precursor by sequence similarity with a human protein, Rpp20, which rRNAs at the A3 site in the internal transcribed sequence of copurifies with human RNase P. Epitope-tagged Rpp2 can be the 35S precursor rRNA (21) and also cleaves RNA primers found in association with both RNase P and RNase mitochon- for mitochondrial DNA replication (22, 23). Although RNase drial RNA processing in immunoprecipitates from crude MRP does not cleave ptRNAs in vitro, its role in rRNA extracts of cells. Depletion of Rpp2 protein in vivo causes processing is affected by proteins that associate with RNase P accumulation of precursor tRNAs with unprocessed introns in vivo (11–14). RNase P functions in the biosynthesis of and 5* and 3* termini, and leads to defects in the processing tRNAs (8) and appears to have a role in rRNA processing in of the 35S precursor rRNA. -

Role and Regulation of the P53-Homolog P73 in the Transformation of Normal Human Fibroblasts
Role and regulation of the p53-homolog p73 in the transformation of normal human fibroblasts Dissertation zur Erlangung des naturwissenschaftlichen Doktorgrades der Bayerischen Julius-Maximilians-Universität Würzburg vorgelegt von Lars Hofmann aus Aschaffenburg Würzburg 2007 Eingereicht am Mitglieder der Promotionskommission: Vorsitzender: Prof. Dr. Dr. Martin J. Müller Gutachter: Prof. Dr. Michael P. Schön Gutachter : Prof. Dr. Georg Krohne Tag des Promotionskolloquiums: Doktorurkunde ausgehändigt am Erklärung Hiermit erkläre ich, dass ich die vorliegende Arbeit selbständig angefertigt und keine anderen als die angegebenen Hilfsmittel und Quellen verwendet habe. Diese Arbeit wurde weder in gleicher noch in ähnlicher Form in einem anderen Prüfungsverfahren vorgelegt. Ich habe früher, außer den mit dem Zulassungsgesuch urkundlichen Graden, keine weiteren akademischen Grade erworben und zu erwerben gesucht. Würzburg, Lars Hofmann Content SUMMARY ................................................................................................................ IV ZUSAMMENFASSUNG ............................................................................................. V 1. INTRODUCTION ................................................................................................. 1 1.1. Molecular basics of cancer .......................................................................................... 1 1.2. Early research on tumorigenesis ................................................................................. 3 1.3. Developing -

Nuclear Domains of the RNA Subunit of Rnase P
Journal of Cell Science 110, 829-837 (1997) 829 Printed in Great Britain © The Company of Biologists Limited 1997 JCS8126 Nuclear domains of the RNA subunit of RNase P Marty R. Jacobson1, Long-Guang Cao1,*, Krishan Taneja2, Robert H. Singer2,†, Yu-li Wang1 and Thoru Pederson1,‡ 1Cell Biology Group, Worcester Foundation for Biomedical Research, Shrewsbury, MA 01545, USA 2Department of Cell Biology, University of Massachusetts Medical Center, Worcester, MA 01655, USA *Present address: Department of Molecular Genetics and Microbiology, University of Florida, PO Box 100266, Gainesville, FL 32610, USA †Present address: Department of Anatomy and Structural Biology, Albert Einstein College of Medicine, Bronx, NY 10461-1975, USA ‡Author for correspondence (e-mail: [email protected]) SUMMARY The ribonucleoprotein enzyme RNase P catalyzes the 5′ microinjection. In contrast, a truncated RNase P RNA con- processing of pre-transfer RNA, and has also recently been taining the To binding domain but lacking nucleotides 89- implicated in pre-ribosomal RNA processing. In the 341 became rapidly localized in nucleoli following nuclear present investigation, in situ hybridization revealed that microinjection. However, unlike the full-length RNase P RNase P RNA is present throughout the nucleus of RNA, this 3′ truncated RNA remained stably associated mammalian cells. However, rhodamine-labeled human with the nucleoli and did not translocate to the nucleo- RNase P RNA microinjected into the nucleus of rat kidney plasm. These results suggest a nucleolar phase in the mat- (NRK) epithelial cells or human (HeLa) cells initially uration, ribonucleoprotein assembly or function of RNase localized in nucleoli, and subsequently became more evenly P RNA, mediated at least in part by the nucleolar To distributed throughout the nucleus, similar to the steady- antigen. -

Accurate Prediction of Kinase-Substrate Networks Using
bioRxiv preprint doi: https://doi.org/10.1101/865055; this version posted December 4, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Accurate Prediction of Kinase-Substrate Networks Using Knowledge Graphs V´ıtNov´aˇcek1∗+, Gavin McGauran3, David Matallanas3, Adri´anVallejo Blanco3,4, Piero Conca2, Emir Mu~noz1,2, Luca Costabello2, Kamalesh Kanakaraj1, Zeeshan Nawaz1, Sameh K. Mohamed1, Pierre-Yves Vandenbussche2, Colm Ryan3, Walter Kolch3,5,6, Dirk Fey3,6∗ 1Data Science Institute, National University of Ireland Galway, Ireland 2Fujitsu Ireland Ltd., Co. Dublin, Ireland 3Systems Biology Ireland, University College Dublin, Belfield, Dublin 4, Ireland 4Department of Oncology, Universidad de Navarra, Pamplona, Spain 5Conway Institute of Biomolecular & Biomedical Research, University College Dublin, Belfield, Dublin 4, Ireland 6School of Medicine, University College Dublin, Belfield, Dublin 4, Ireland ∗ Corresponding authors ([email protected], [email protected]). + Lead author. 1 bioRxiv preprint doi: https://doi.org/10.1101/865055; this version posted December 4, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Abstract Phosphorylation of specific substrates by protein kinases is a key control mechanism for vital cell-fate decisions and other cellular pro- cesses. However, discovering specific kinase-substrate relationships is time-consuming and often rather serendipitous. -

Capturing the SARS-Cov-2 RNA Genome and Its Bound Proteins with RAP-MS
SUPPLEMENTARY FIGURES Supplementary Figure 1 | Capturing the SARS-CoV-2 RNA genome anD its bounD proteins with RAP-MS. a, Alignment of protein-crosslinked RNA fragments to the SARS- CoV-2 genome following RNA antisense purification of SARS-CoV-2. Two replicate experiments are shown. b, Fraction of all aligned sequence reads mapping to the human or SARS-CoV-2 genomes in pilot RAP-MS experiment. c, As in b, but for full scale SARS-CoV- 2 RAP-MS and RMRP RAP-MS experiments. D, Western blot of SARS-CoV-2 and RMRP RAP-MS experiments. Antibodies for SARS-CoV-2 nucleoprotein (Abcam, ab272852) and human POP1 (Proteintech, 12029-1-AP) were used. 1 Supplementary Figure 2 | Connecting the SARS-CoV-2 RNA interactome to proteome dynamics in infected cells. a, Principle component analysis for proteome measurements of SARS-CoV-2 infected or Mock infected HuH-7 cells. b, Gene set enrichment analysis for proteins significantly regulated in global proteome measurements. Gene sets enriched in 2 addition to those shown in Figure 3b are presented. c, Protein-protein association network of expanded SARS-CoV-2 interactome proteins (blue: interactome protein, not regulated; red: interactome protein, regulated) and their connections to differentially regulated proteins upon SARS-CoV-2 infection. Upregulated proteins are shown in light grey; downregulated proteins are shown in dark grey. Circle sizes scale to the number of connections of each interactome protein. 3 Supplementary Figure 3 | Functional valiDation of SARS-CoV-2-binding proteins. a, Schematic of dual-fluorescence translation reporter to quantify ribosomal frameshifting efficiency. The depicted control construct contains EGFP and mCherry in an in-frame orientation, leading to the production of both fluorescence proteins separated by a self- cleaving 2A peptide when the 0 reading frame is translated. -

Cryo-EM Structure of Catalytic Ribonucleoprotein Complex Rnase MRP
bioRxiv preprint doi: https://doi.org/10.1101/2020.03.17.996132; this version posted March 18, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Cryo-EM structure of catalytic ribonucleoprotein complex RNase MRP Authors: Anna Perederina1, Di Li1, Hyunwook Lee1, Carol Bator1, Igor Berezin1, Susan L. Hafenstein1,2, Andrey S. Krasilnikov1,3* Affiliations: 1Department of Biochemistry and Molecular Biology, Pennsylvania State University, University Park, PA 16802 2Department of Medicine, Pennsylvania State University, Hershey, PA 17033 3Center for RNA Biology, Pennsylvania State University, University Park, PA 16802 *Correspondence to: [email protected] Abstract: RNase MRP is an essential eukaryotic ribonucleoprotein complex involved in the maturation of rRNA and the regulation of the cell cycle. RNase MRP is related to the ribozyme-based RNase P, but it has evolved to have distinct cellular roles. We report a cryo-EM structure of the S. cerevisiae RNase MRP holoenzyme solved to 3.0 Å. We describe the structure of this 450 kDa complex, interactions between its components, and the organization of its catalytic RNA. We show that while the catalytic center of RNase MRP is inherited from the ancestral enzyme RNase P, the substrate binding pocket of RNase MRP is significantly altered by the addition of unique RNA and protein elements, as well as by RNA-driven protein remodeling. One Sentence Summary: Changes in peripheral RNA elements and RNA-driven protein remodeling result in diversification of related catalytic RNPs 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.03.17.996132; this version posted March 18, 2020. -
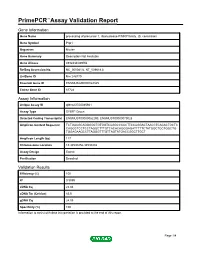
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name processing of precursor 1, ribonuclease P/MRP family, (S. cerevisiae) Gene Symbol Pop1 Organism Mouse Gene Summary Description Not Available Gene Aliases 4932434G09Rik RefSeq Accession No. NC_000081.6, NT_039618.8 UniGene ID Mm.248779 Ensembl Gene ID ENSMUSG00000022325 Entrez Gene ID 67724 Assay Information Unique Assay ID qMmuCED0045941 Assay Type SYBR® Green Detected Coding Transcript(s) ENSMUST00000052290, ENSMUST00000079028 Amplicon Context Sequence TCTGACGCAGGGGCTGTGGTCAGGCCCCCTTCCAGGACTAACCTCACACTGCTC CAGGCTCCTCCTAGGCTTTGTCACACAGGGAGATTTTTCTATGGCTGCTGGCTG TGGAGAAGCCTTAGGGTTTGTTAGTATGACCGGCTTGCT Amplicon Length (bp) 117 Chromosome Location 15:34530256-34530402 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 100 R2 0.9999 cDNA Cq 23.04 cDNA Tm (Celsius) 83.5 gDNA Cq 24.05 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 1/4 PrimePCR™Assay Validation Report Pop1, Mouse Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 2/4 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Mouse Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. Detected Coding Transcript(s) This is a list of the Ensembl transcript ID(s) that this assay will detect. -

The SARS-Cov-2 RNA–Protein Interactome in Infected Human Cells
The SARS-CoV-2 RNA–protein interactome in infected human cells The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Schmidt, Nora et al. "The SARS-CoV-2 RNA–protein interactome in infected human cells." Nature Microbiology (December 2020): doi.org/10.1038/s41564-020-00846-z. © 2020 The Author(s) As Published http://dx.doi.org/10.1038/s41564-020-00846-z Publisher Springer Science and Business Media LLC Version Final published version Citable link https://hdl.handle.net/1721.1/128950 Terms of Use Creative Commons Attribution 4.0 International license Detailed Terms https://creativecommons.org/licenses/by/4.0/ ARTICLES https://doi.org/10.1038/s41564-020-00846-z The SARS-CoV-2 RNA–protein interactome in infected human cells Nora Schmidt 1,10, Caleb A. Lareau 2,10, Hasmik Keshishian3,10, Sabina Ganskih 1, Cornelius Schneider4,5, Thomas Hennig6, Randy Melanson3, Simone Werner1, Yuanjie Wei1, Matthias Zimmer1, Jens Ade 1, Luisa Kirschner6, Sebastian Zielinski 1, Lars Dölken1,6, Eric S. Lander3,7,8, Neva Caliskan1,9, Utz Fischer1,5, Jörg Vogel 1,4, Steven A. Carr3, Jochen Bodem 6 ✉ and Mathias Munschauer 1 ✉ Characterizing the interactions that SARS-CoV-2 viral RNAs make with host cell proteins during infection can improve our understanding of viral RNA functions and the host innate immune response. Using RNA antisense purification and mass spec- trometry, we identified up to 104 human proteins that directly and specifically bind to SARS-CoV-2 RNAs in infected human cells. We integrated the SARS-CoV-2 RNA interactome with changes in proteome abundance induced by viral infection and linked interactome proteins to cellular pathways relevant to SARS-CoV-2 infections. -

An Integrative Genomic Analysis of the Longshanks Selection Experiment for Longer Limbs in Mice
bioRxiv preprint doi: https://doi.org/10.1101/378711; this version posted August 19, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Title: 2 An integrative genomic analysis of the Longshanks selection experiment for longer limbs in mice 3 Short Title: 4 Genomic response to selection for longer limbs 5 One-sentence summary: 6 Genome sequencing of mice selected for longer limbs reveals that rapid selection response is 7 due to both discrete loci and polygenic adaptation 8 Authors: 9 João P. L. Castro 1,*, Michelle N. Yancoskie 1,*, Marta Marchini 2, Stefanie Belohlavy 3, Marek 10 Kučka 1, William H. Beluch 1, Ronald Naumann 4, Isabella Skuplik 2, John Cobb 2, Nick H. 11 Barton 3, Campbell Rolian2,†, Yingguang Frank Chan 1,† 12 Affiliations: 13 1. Friedrich Miescher Laboratory of the Max Planck Society, Tübingen, Germany 14 2. University of Calgary, Calgary AB, Canada 15 3. IST Austria, Klosterneuburg, Austria 16 4. Max Planck Institute for Cell Biology and Genetics, Dresden, Germany 17 Corresponding author: 18 Campbell Rolian 19 Yingguang Frank Chan 20 * indicates equal contribution 21 † indicates equal contribution 22 Abstract: 23 Evolutionary studies are often limited by missing data that are critical to understanding the 24 history of selection. Selection experiments, which reproduce rapid evolution under controlled 25 conditions, are excellent tools to study how genomes evolve under strong selection. Here we 1 bioRxiv preprint doi: https://doi.org/10.1101/378711; this version posted August 19, 2018. -

Identification of Key Proteins Involved in Axon Guidance Related Disorders: a Systems Biology Approach
Identification of Key Proteins Involved in Axon Guidance Related Disorders: A Systems Biology Approach Ishtiaque Ahammad1* 1. Department of Biochemistry and Microbiology, North South University, Dhaka, Bangladesh. * Corresponding author. Email: [email protected] Abstract Axon guidance is a crucial process for growth of the central and peripheral nervous systems. In this study, 3 axon guidance related disorders, namely- Duane Retraction Syndrome (DRS) , Horizontal Gaze Palsy with Progressive Scoliosis (HGPPS) and Congenital fibrosis of the extraocular muscles type 3 (CFEOM3) were studied using various Systems Biology tools to identify the genes and proteins involved with them to get a better idea about the underlying molecular mechanisms including the regulatory mechanisms. Based on the analyses carried out, 7 significant modules have been identified from the PPI network. Five pathways/processes have been found to be significantly associated with DRS, HGPPS and CFEOM3 associated genes. From the PPI network, 3 have been identified as hub proteins- DRD2, UBC and CUL3. Keywords Axon Guidance ; DRS ; HGPPS ; CFEOM3 ; in silico ; Systems Biology ; PPI network Introduction Duane Retraction Syndrome (DRS) is a human genetic disorder of cranial nerve guidance (Engle 2010). It’s a disorder of ocular motility involving deficient horizontal eye movements, eyelid retraction, palpebral fissure narrowing in adduction and a variety of other abnormal movement of the affected eye when the other eye is fixated in various cardinal positions (SHALABY and BAHGAT 2010; ALEXANDRAKIS and SAUNDERS 2001) . The absence of 6th cranial nerve and the consequent developmental adaptation that occurs in the embryo results in the abnormal pattern of ocular motility of DRS (SHALABY and BAHGAT 2010). -

Cryo-EM Structure of Catalytic Ribonucleoprotein Complex Rnase MRP
ARTICLE https://doi.org/10.1038/s41467-020-17308-z OPEN Cryo-EM structure of catalytic ribonucleoprotein complex RNase MRP Anna Perederina1,DiLi1, Hyunwook Lee1, Carol Bator1, Igor Berezin1, Susan L. Hafenstein1,2 & ✉ Andrey S. Krasilnikov 1,3 RNase MRP is an essential eukaryotic ribonucleoprotein complex involved in the maturation of rRNA and the regulation of the cell cycle. RNase MRP is related to the ribozyme-based 1234567890():,; RNase P, but it has evolved to have distinct cellular roles. We report a cryo-EM structure of the S. cerevisiae RNase MRP holoenzyme solved to 3.0 Å. We describe the structure of this 450 kDa complex, interactions between its components, and the organization of its catalytic RNA. We show that some of the RNase MRP proteins shared with RNase P undergo an unexpected RNA-driven remodeling that allows them to bind to divergent RNAs. Further, we reveal how this RNA-driven protein remodeling, acting together with the introduction of new auxiliary elements, results in the functional diversification of RNase MRP and its progenitor, RNase P, and demonstrate structural underpinnings of the acquisition of new functions by catalytic RNPs. 1 Department of Biochemistry and Molecular Biology, Pennsylvania State University, University Park, 16802 PA, USA. 2 Department of Medicine, Pennsylvania ✉ State University, Hershey, 17033 PA, USA. 3 Center for RNA Biology, Pennsylvania State University, University Park, 16802 PA, USA. email: [email protected] NATURE COMMUNICATIONS | (2020) 11:3474 | https://doi.org/10.1038/s41467-020-17308-z | www.nature.com/naturecommunications 1 ARTICLE NATURE COMMUNICATIONS | https://doi.org/10.1038/s41467-020-17308-z ibonuclease (RNase) MRP, a site-specific endoribonuclease, RNases P have been determined21,22.