Development of Efficiently Produced, Renewable Polycarbonates from Fatty Acids, CO2, and Propylene Oxide for Plastic Film Applications
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Polycarbonate Lenses
Polycarbonate Lenses The most impact resistant of all lens materials is polycarbonate. Children, athletes, anyone working at a job or hobby where they might get hit in the face and need safety glasses, people with only one eye, those who fall a lot, are natural candidates for polycarbonate lenses. If safety is a prime concern, choose polycarbonate lenses. Advantages of polycarbonate lenses : 1. Polycarbonate has four to five times the impact resistance of glass or plastic. When glass or plastic lenses break, they do not break into harmless granules, but can break into sharp shards that can enter your eye and destroy your vision. Poly- carbonate is far and away the safest of all the lenses made. 2. Polycarbonate is the lightest lens material made. 3. Polycarbonate lenses naturally provide protection against ultra-violet light, at no additional charge. 4. Polycarbonate lenses come with a scratch resistant coating (not scratch proof) at no additional charge. 5. Polycarbonate is a high index material, so the lenses will be thinner than if made with glass or plastic. Disadvantages of polycarbonate lenses : 1. People in prescriptions with higher powers sometimes have trouble seeing out the edges of the lenses--your clear field of vision is not as wide as with glass or plas- tic lenses. The lenses are made with different curves than are used to make the same pre- scription power in glass or plastic, so you will see out of these lenses a little dif- ferently. People with prescriptions up to plus or minus three diopters (most people) usually have no problem adjusting to polycarbonate lenses. -

(12) Patent Application Publication (10) Pub. No.: US 2011/0309539 A1 Steinke Et Al
US 2011 0309539A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2011/0309539 A1 Steinke et al. (43) Pub. Date: Dec. 22, 2011 (54) METHOD FOR PRODUCING POLYMER Publication Classification MIXTURES (51) Int. Cl. (75) Inventors: Tobias Heinz Steinke, Speyer B29B 9/10 (2006.01) (DE); Hans-Helmut Görtz, (52) U.S. Cl. ............................................................ 264/5 Freinsheim (DE); Jirgen Ahlers, (57) ABSTRACT Gross-Rohrheim (DE); Freddy The present invention relates to a process for the production Gruber, Offenbach (DE); Gabriel of polymer mixtures of i) polypropylene carbonate and ii) at Skupin, Speyer (DE) least one further polymer, including the following steps: (a) reaction of propylene carbonate with carbon dioxide in the (73) Assignee: BASF SE, Ludwigshafen (DE) presence of a Zinc catalyst, cobalt catalyst, or lanthanoid catalyst—in excess propylene carbonate or in an aprotic (21) Appl. No.: 13/254,086 non-water-miscible solvent, PCT Fled: Mar. 1, 2010 (b) addition of an aqueous acidic solution to the reaction (22) mixture after termination of the reaction, (c) removal of the aqueous phase, (86) PCT NO.: PCT/EP2010/052528 (d) optionally washing of the remaining organic phase with S371 (c)(1), Water, (2), (4) Date: Sep. 7, 2011 (e) addition of polymer component ii), (f) degassing and drying of the resultant polymer mixture and (30) Foreign Application Priority Data optionally removal of the aprotic, non-water-miscible sol vent, and Mar. 3, 2009 (EP) .................................. O91542O1.9 (g) pelletization of the polymer melt. US 2011/0309539 A1 Dec. 22, 2011 METHOD FOR PRODUCING POLYMER the polypropylene carbonate itself. The processes described MIXTURES in the literature for the production of polypropylene carbon ate mixtures use mixing of the pellets of the individual com ponents, and then melting of the pellet mixture in an extruder, 0001. -

Migration of Bisphenol a from Polycarbonate Plastic of Different Qualities
Migration of bisphenol A from polycarbonate plastic of different qualities Environmental project No. 1710, 2015 [Series Title and year] Title: Editing: Migration of Bisphenol A from polycarbonate Gitte Alsing Pedersen, DTU National Food Institute, plastic of different qualities Søren Hvilsted, DTU Danish Polymer Centre, Department of Chemical and Biochemical Engineering and Jens Højslev Petersen, DTU National Food Institute Technical University of Denmark Published by: The Danish Environmental Protection Agency Strandgade 29 1401 Copenhagen K Denmark www.mst.dk/english Year: ISBN no. 2015 978-87-93352-24-7 Disclaimer: When the occasion arises, the Danish Environmental Protection Agency will publish reports and papers concerning research and development projects within the environmental sector, financed by study grants provided by the Danish Environmental Protection Agency. It should be noted that such publications do not necessarily reflect the position or opinion of the Danish Environmental Protection Agency. However, publication does indicate that, in the opinion of the Danish Environmental Protection Agency, the content represents an important contribution to the debate surrounding Danish environmental policy. Sources must be acknowledged. 2 Migration of Bisphenol A from polycarbonate plastic of different qualities Contents Foreword .................................................................................................................. 5 Conclusion and Summary ......................................................................................... -

(12) United States Patent (10) Patent No.: US 8,871,053 B2 Sealey Et Al
USOO887 1053B2 (12) United States Patent (10) Patent No.: US 8,871,053 B2 Sealey et al. (45) Date of Patent: Oct. 28, 2014 (54) FIRE RETARDANT TREATED FLUFF PULP USPC .................. 162/9, 141, 149, 158, 159, 164.1, WEB 162/164.6, 168.1-168.2, 166, 183-185, (71) Applicant: International Paper Company, 162/1811-181.2: 8/116.1; 428/212,340, 428/920 Memphis, TN (US) See application file for complete search history. (72) Inventors: James E. Sealey, Anderson, SC (US); Brent A. Fields, Trenton, OH (US) (56) References Cited (73) Assignee: International Paper Company, Memphis, TN (US) U.S. PATENT DOCUMENTS (*) Notice: Subject to any disclaimer, the term of this 1,382,618 A 6, 1921 Blenio patent is extended or adjusted under 35 2,654,295 A 10, 1953 Sutherland U.S.C. 154(b) by 0 days. (Continued) (21) Appl. No.: 14/192,920 FOREIGN PATENT DOCUMENTS (22) Filed: Feb. 28, 2014 EP O132128 1, 1985 (65) Prior Publication Data GB 2209352 5, 1989 US 2014/O174681 A1 Jun. 26, 2014 (Continued) Related U.S. Application Data OTHER PUBLICATIONS (60) Division of application No. 13/751,218, filed on Jan. Flame Retardants for Plastics and texiles, Weil, et al., Hanser Pub 28, 2013, now Pat. No. 8,685,206, which is a lishers, Munich 2009, p. 4-8. continuation of application No. PCT/US2011/046173, filed on Aug. 2, 2011. (Continued) (60) Provisional application No. 61/370.236, filed on Aug. Primary Examiner — Jose Fortuna 3, 2010. (74) Attorney, Agent, or Firm — Thomas W. -

Blends of Polycarbonate Containing Fluorinated-Bisphenol-A and Polyvinyl Chloride
Europaisches Patentamt European Patent Office © Publication number: 0 576 057 A1 Office europeen des brevets EUROPEAN PATENT APPLICATION © Application number: 93201533.2 int. Ci.5; C08L 69/00, C08L 27/06, C08G 64/10, //(C08L69/00, @ Date of filing: 28.05.93 27:06),(C08L27/06,69:00) © Priority: 01.06.92 US 891032 © Applicant: ENICHEM S.p.A. Piazza della Repubblica, 16 @ Date of publication of application: 1-20124 Milano(IT) 29.12.93 Bulletin 93/52 @ Inventor: Drzewinski, Michael A. © Designated Contracting States: 371 Clarksville Road, Princeton Junction AT BE CH DE DK ES FR GB GR IE IT LI LU MC New Jersey 08850(US) NL PT SE © Representative: Roggero, Sergio et al Ing. Barzano & Zanardo Milano S.p.A. Via Borgonuovo 10 1-20121 Milano (IT) © Blends of polycarbonate containing fluorinated-bisphenol-A and polyvinyl chloride. © Bisphenol A polycarbonate containing at least 15 mole % of 2,2-bis-(4-hydroxyphenyl)hexafluoropropane (6F-Bisphenol A) can be blended with polyvinyl chloride (PVC) to form a thermodynamically miscible, transpar- ent, single phase blend at all compositions. Such blends are flame resistant as well as resistant to attack by acids, bases and many organic solvents. CO Rank Xerox (UK) Business Services (3. 10/3.6/3.3. 1) EP 0 576 057 A1 BACKGROUND OF THE INVENTION Field of the Invention: 5 This invention pertains to mixtures of polyvinyl chloride (PVC) and polycarbonates which contain at least 15 mole % of fluorinated bisphenol monomer units (F-PC) such as 2,2-bis-(4-hydroxyphenyl)- hexafluoropropane (6F-bisphenol A), herein referred to as 6F-PC. -

The Asymmetric Synthesis of Styrene-Oxide and Its Reactions with Dialkylmagnesium Reagents and Boron Hydrides
University of New Hampshire University of New Hampshire Scholars' Repository Doctoral Dissertations Student Scholarship Summer 1971 THE ASYMMETRIC SYNTHESIS OF STYRENE-OXIDE AND ITS REACTIONS WITH DIALKYLMAGNESIUM REAGENTS AND BORON HYDRIDES RONALD LEROY ATKINS Follow this and additional works at: https://scholars.unh.edu/dissertation Recommended Citation ATKINS, RONALD LEROY, "THE ASYMMETRIC SYNTHESIS OF STYRENE-OXIDE AND ITS REACTIONS WITH DIALKYLMAGNESIUM REAGENTS AND BORON HYDRIDES" (1971). Doctoral Dissertations. 964. https://scholars.unh.edu/dissertation/964 This Dissertation is brought to you for free and open access by the Student Scholarship at University of New Hampshire Scholars' Repository. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of University of New Hampshire Scholars' Repository. For more information, please contact [email protected]. 72-3736 ATKINS, Ronald Leroy, 1939- THE ASYMMETRIC SYNTHESIS OF STYRENE OXIDE AND ITS REACTIONS WITH DIALKYLMAGNESIUM REAGENTS AND BORON HYDRIDES. University of New Hampshire, Ph.D., 1971 Chemistry, organic University Microfilms, A XEROX Company, Ann Arbor, Michigan © 1971 Ronald LeRoy Atkina ALL RIGHTS RESERVED THE ASYMMETRIC SYNTHESIS OF STYRENE OXIDE AND ITS REACTIONS WITH DIALKYLMAGNESIUM REAGENTS AND BORON HYDRIDES by RONALD L. ATKINS B. S., The University of Wyoming, 1966 M. S., The University of Wyoming, 1968 A THESIS Submitted to the University of New Hampshire In Partial Fulfillment of The Requirements for the Degree of DOCTOR OF PHILOSOPHY Graduate School Department of Chemistry August, 1971 This thesis has been examined and approved. f b w < M & > YUffvyido— The^js Director, James D. Morrison Asspaiate Professor of Chemistry & L DJitdsh-iU. _____ Colin D. -

Study of Surface Mechanical Characteristics of ABS/PC Blends Using Nanoindentation
processes Article Study of Surface Mechanical Characteristics of ABS/PC Blends Using Nanoindentation Saira Bano 1, Tanveer Iqbal 2, Naveed Ramzan 3 and Ujala Farooq 4,* 1 Department of Chemical & Polymer Engineering, University of Engineering & Technology, FSD Campus, Lahore 38000, Pakistan; [email protected] 2 Department of Chemical, Polymer & Composite Materials Engineering, University of Engineering & Technology, KSK Campus, Lahore 54890, Pakistan; [email protected] 3 Department of Chemical Engineering, University of Engineering & Technology, KSK Campus, Lahore 54890, Pakistan; [email protected] 4 Faculty of Aerospace Engineering, Aerospace Manufacturing Technologies, Delft University of Technology, Kluyverweg 1, 2629 HS Delft, The Netherlands * Correspondence: [email protected] Abstract: Acrylonitrile butadiene styrene (ABS) and polycarbonate (PC) are considered a well-known class of engineering thermoplastics due to their efficient use in automotive, 3D printing, and elec- tronics. However, improvement in toughness, processability, and thermal stability is achieved by mixing together ABS and PC. The present study focuses on the understanding of surface mechani- cal characterization of acrylonitrile butadiene styrene (ABS) and polycarbonate (PC) blends using nano-indentation. Polymer blends sheets with three different proportions of ABS/PC (75:25, 50:50, and 25:75) were fabricated via melt-processing and thermal press. Fourier transform infrared (FTIR) spectroscopy was performed to analyze the intermolecular interactions between the blends’ compo- nents. To understand the surface mechanical properties of ABS and PC blends, a sufficient number Citation: Bano, S.; Iqbal, T.; Ramzan, of nano-indentation tests were performed at a constant loading rate to a maximum load of 100 mN. N.; Farooq, U. -

Polycarbonate (PC)
Polycarbonate (PC) Carlos Buitrago Introduction to Polymers CE 435 OVERVIEW zIntroduction zStructure of PC zSynthesis zManufacturing zPhysical Properties of PC zApplications of PC zPC Blends zQuestions INTRODUCTION z Polycarbonate (PC), was first developed in 1953 by Bayer in Germany, and General Electric in the US independently. Its most popular trade name is LEXAN® z PC is one of the high performance heterochain polymeric materials that comprise the family of “engineering thermoplastics” z PC is a good material choice in industry not only due to its characteristics, but also because its processing is environmentally friendly, and it can be recycled STRUCTURE OF PC z A polycarbonate molecule is composed by a Bisphenol A part and a carbonate group z Bisphenol A contains two aromatic rings, which are responsible for PC’s stiff LG-DOW (n.d.). Molecular structure of polycarbonate. backbone http://www.lg-dow.com/tech/tech.htm z The Bisphenol A group also • The Characteristic high contributes to PC’s inability glass transition temperature to crystallize. This (Tg = 145ºC) of PC is caused amorphous structure gives by the minimal molecular the polymer its particular rotation about the bonds transparency SYNTHESIS PC is most often synthesized from Bisphenol A and phosgene by a step-growth polymerization in which Cl- ions are eliminated every time the monomers react. This kind of step-growth polymerization is often called a condensation process Polymerization Steps 1. The Bisphenol A groups are reacted with proton acceptors such as NaOH to obtain the polymerization functional groups Polymerization steps (Cont’d) 2. The deprotonated Bispohenol A reacts with Phosgene and a catalyst at temperatures between 25 and 35ºC. -

Characterization of High Molecular Weight Poly(Vinyl Chloride) – Lithium Tetraborate Electrolyte Plasticized by Propylene Carbonate
9 Characterization of High Molecular Weight Poly(vinyl chloride) – Lithium Tetraborate Electrolyte Plasticized by Propylene Carbonate Ramesh T. Subramaniam1,*, Liew Chiam-Wen1, Lau Pui Yee2 and Ezra Morris2 1Centre for Ionics University Malaya, Department of Physics, Faculty of Science, University of Malaya, Kuala Lumpur 2Faculty of Engineering and Science, Universiti Tunku Abdul Rahman, Kuala Lumpur Malaysia 1. Introduction An electrolyte is a substance consisting of free ions and acts as a medium channel for transferring the charges between a pair of electrodes. Sometimes, they are also referred as “lytes” which is derived from the Greek word, “lytos”, which means “it may be dissolved”. Electrolyte is comprised of positively charged species which is called as cation and negatively charged species, anion. The properties of an electrolyte can be exploited via electrolysis process: separation of chemically bonded element or compounds by applying the electrical current. In the early stage, liquid electrolytes have been discovered and investigated. Liquid electrolyte is a substance that conducts the electricity in an aqueous solution by migrating both cations and anions to the opposite electrodes through an electrically conducting path as a useful electric current. However, it faces problem of leakage of hazardous liquids or gases. Other drawbacks are formation of lithium dendrite, electrolytic degradation of electrolyte and uses of flammable organic solvent (Ramesh et al., 2011a). Apart from that, it exhibits poor long–term stability due to the evaporation of the liquid phase in the cells (Yang et al., 2008). Therefore, solid polymer electrolytes (SPEs) were synthesized to prevail over the limitations of conventional liquid electrolytes. 1.1 Solid polymer electrolytes The development on SPEs was initiated by the pioneering work of Wright et al. -
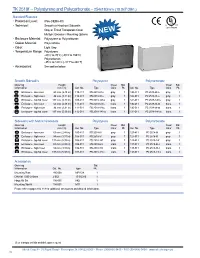
TK 2518F – Polystyrene and Polycarbonate – 254X180mm (10.0X7.09In.)
TK 2518f – Polystyrene and Polycarbonate – 254x180mm (10.0x7.09in.) Standard Features • Protection Level: IP66 (NEMA 4X) • Technical: Smooth or Knockout Sidewalls Upon Request Gray or Tinted Transparent Cover ➀ Multiple Enclosure Mounting Options NEW • Enclosure Material: Polystyrene or Polycarbonate • Gasket Material: Polyurethane • Color: Light Gray • Temperature Range: Polystyrene -40°C to 70°C (-40°F to 158°F) Polycarbonate -35°C to 120°C (-31°F to 248°F) • Accessories: See section below Smooth Sidewalls Polystyrene Polycarbonate Ordering Height Cover Std. Cover Std. Information mm (in) Cat. No. Type Color Pk. Cat. No. Type Color Pk. Enclosure - low cover 63 mm (2.48 in) 110-411 PS 2518-6f-o gray 1 120-411 PC 2518-6f-o gray 1 Enclosure - high cover 84 mm (3.31 in) 110-911 PS 2518-8f-o gray 1 120-911 PC 2518-8f-o gray 1 Enclosure - top hat cover 137 mm (5.39 in) 109-411 PS 2518-14f-o gray 1 121-411 PC 2518-14f-o gray 1 Enclosure - low cover 63 mm (2.48 in) 111-011 PS 2518-6f-to trans. 1 130-011 PC 2518-6f-to trans. 1 Enclosure - high cover 84 mm (3.31 in) 111-511 PS 2518-8f-to trans. 1 130-511 PC 2518-8f-to trans. 1 Enclosure - top hat cover 137 mm (5.39 in) 112-011 PS 2518-14f-to trans. 1 131-011 PC 2518-14f-to trans. 1 Sidewalls with Metric Knockouts Polystyrene Polycarbonate Ordering Height Cover Std. Cover Std. Information mm (in) Cat. No. Type Color Pk. -

Applicability of Electrospun Polypropylene Carbonate Polymers As a Drug Carrier for Sirolimus
MOLECULAR MEDICINE REPORTS 15: 4253-4258, 2017 Applicability of electrospun polypropylene carbonate polymers as a drug carrier for sirolimus HOURONG SUN, XINGHUA GU, KAI LIU, CHANGCUN FANG and MENGMENG TANG Department of Cardiovascular Surgery, Qilu Hospital, Shandong University, Jinan, Shandong 250012, P.R. China Received April 6, 2016; Accepted March 8, 2017 DOI: 10.3892/mmr.2017.6540 Abstract. Polypropylene carbonate (PPC), a biodegradable Introduction aliphatic polyester, exhibits one particular advantage over other polyesters, which is that following degradation in vivo, Polymers may be naturally-occurring, including collagen it primarily produces H2O and CO2, causing minimal side and polysaccharides, or synthesized, including polylac- effects. Although PPC exhibits limited mechanical strength, tide (PLA), polyethylene and poly lactic-co-glycolic acid and is therefore not able to serve as a scaffold to support tissue (PLGA) (1-7). Polymers have been extensively studied and regeneration, it may be suitable for drug delivery; however, used for biomedical and pharmaceutical applications (1,8). this requires further investigation. In the present study, elec- As polymer synthesis may be conducted under controlled trospinning was applied to generate PPC polymers containing conditions, and the properties of polymers, including hydro- sirolimus, a cell growth-inhibiting drug which is used to treat philicity, degradability and biocompatibility, may be tailored restenosis. The properties of PPC-sirolimus polymers were for specific applications, numerous studies have focused on examined using scanning electron microscopy, differential the development of novel synthetic polymers for tissue-engi- scanning calorimetry and in vitro degradation assays. Drug neering (polymers as scaffolds for tissue regeneration) and loading and entrapment efficiency were determined, and controlled drug release (polymers as drug carriers) (3,4,9). -

Retropath2.0: a Retrosynthesis Workflow for Metabolic Engineers
bioRxiv preprint doi: https://doi.org/10.1101/141721; this version posted June 29, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. RetroPath2.0: a retrosynthesis workflow for metabolic engineers Baudoin Delépine1,2,3,§, Thomas Duigou3,§, Pablo Carbonell4, Jean-Loup Faulon3,1,2,4,* 1 CNRS-UMR8030 / Laboratoire iSSB, Université Paris-Saclay, Évry 91000, France 2 CEA, DRF, IG, Genoscope, Évry 91000, France 3 Micalis Institute, INRA, AgroParisTech, Université Paris-Saclay, 78350 Jouy-en-Josas, France 4 SYNBIOCHEM Centre, Manchester Institute of Biotechnology, University of Manchester, Manchester, UK §: These authors contributed equally to this work. *: Corresponding author at: Micalis, Institut National de la Recherche Agronomique, Domaine de Vilvert, 78352 JOUY-EN-JOSAS, FRANCE; E-mail address: [email protected] Highlights • State-of-the-art Computer-Aided Design retrosynthesis solutions lack open source and ease of use • We propose RetroPath2.0 a modular and open-source workflow to perform retrosynthesis • RetroPath2.0 computes reaction network between Source and Sink sets of compounds • RetroPath2.0 is distributed as a KNIME workflow for desktop computers • RetroPath2.0 is ready-for-use and distributed with reaction rules Funding This work was supported by the French National Research Agency [ANR-15-CE1-0008], the Biotechnology and Biological Sciences Research Council, Centre for synthetic biology of fine and speciality chemicals [BB/M017702/1]; Synthetic Biology Applications for Protective Materials [EP/N025504/1], and GIP Genopole.