Modelling Mitochondrial Disease in Human Pluripotent Stem Cells: What Have We Learned?
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Establishing the Pathogenicity of Novel Mitochondrial DNA Sequence Variations: a Cell and Molecular Biology Approach
Mafalda Rita Avó Bacalhau Establishing the Pathogenicity of Novel Mitochondrial DNA Sequence Variations: a Cell and Molecular Biology Approach Tese de doutoramento do Programa de Doutoramento em Ciências da Saúde, ramo de Ciências Biomédicas, orientada pela Professora Doutora Maria Manuela Monteiro Grazina e co-orientada pelo Professor Doutor Henrique Manuel Paixão dos Santos Girão e pela Professora Doutora Lee-Jun C. Wong e apresentada à Faculdade de Medicina da Universidade de Coimbra Julho 2017 Faculty of Medicine Establishing the pathogenicity of novel mitochondrial DNA sequence variations: a cell and molecular biology approach Mafalda Rita Avó Bacalhau Tese de doutoramento do programa em Ciências da Saúde, ramo de Ciências Biomédicas, realizada sob a orientação científica da Professora Doutora Maria Manuela Monteiro Grazina; e co-orientação do Professor Doutor Henrique Manuel Paixão dos Santos Girão e da Professora Doutora Lee-Jun C. Wong, apresentada à Faculdade de Medicina da Universidade de Coimbra. Julho, 2017 Copyright© Mafalda Bacalhau e Manuela Grazina, 2017 Esta cópia da tese é fornecida na condição de que quem a consulta reconhece que os direitos de autor são pertença do autor da tese e do orientador científico e que nenhuma citação ou informação obtida a partir dela pode ser publicada sem a referência apropriada e autorização. This copy of the thesis has been supplied on the condition that anyone who consults it recognizes that its copyright belongs to its author and scientific supervisor and that no quotation from the -

TRNT1 Gene Trna Nucleotidyl Transferase 1
TRNT1 gene tRNA nucleotidyl transferase 1 Normal Function The TRNT1 gene provides instructions for making a protein involved in the production ( synthesis) of other proteins. During protein synthesis, a molecule called transfer RNA ( tRNA) helps assemble protein building blocks (amino acids) into a chain that forms the protein. Each tRNA carries a specific amino acid to the growing chain. The TRNT1 protein modifies tRNAs by adding a series of three DNA building blocks (nucleotides), called a CCA trinucleotide, to the molecule. This modification is essential for the correct amino acid to be attached to each tRNA. While most protein synthesis occurs in the fluid surrounding the nucleus (cytoplasm), some proteins are synthesized in cell structures called mitochondria, which are the energy-producing centers in cells. Many mitochondrial proteins form groups (complexes) that carry out the reactions that produce energy. Separate tRNA molecules are used to build proteins in the cytoplasm and mitochondria. The TRNT1 protein attaches the CCA trinucleotide to both cytoplasmic and mitochondrial tRNA molecules. Health Conditions Related to Genetic Changes TRNT1 deficiency More than 20 TRNT1 gene mutations have been found to cause TRNT1 deficiency, a condition with a range of signs and symptoms that affect many body systems. Features can include a blood disorder called sideroblastic anemia, recurrent fevers, a shortage of immune cells called B cells that leads to impairment of the immune system ( immunodeficiency), delayed development of speech and motor skills, and eye abnormalities that cause vision problems. The severity of the condition varies among affected individuals. The TRNT1 gene mutations that cause TRNT1 deficiency lead to a shortage (deficiency) of functional TRNT1 protein. -

Table 2. Significant
Table 2. Significant (Q < 0.05 and |d | > 0.5) transcripts from the meta-analysis Gene Chr Mb Gene Name Affy ProbeSet cDNA_IDs d HAP/LAP d HAP/LAP d d IS Average d Ztest P values Q-value Symbol ID (study #5) 1 2 STS B2m 2 122 beta-2 microglobulin 1452428_a_at AI848245 1.75334941 4 3.2 4 3.2316485 1.07398E-09 5.69E-08 Man2b1 8 84.4 mannosidase 2, alpha B1 1416340_a_at H4049B01 3.75722111 3.87309653 2.1 1.6 2.84852656 5.32443E-07 1.58E-05 1110032A03Rik 9 50.9 RIKEN cDNA 1110032A03 gene 1417211_a_at H4035E05 4 1.66015788 4 1.7 2.82772795 2.94266E-05 0.000527 NA 9 48.5 --- 1456111_at 3.43701477 1.85785922 4 2 2.8237185 9.97969E-08 3.48E-06 Scn4b 9 45.3 Sodium channel, type IV, beta 1434008_at AI844796 3.79536664 1.63774235 3.3 2.3 2.75319499 1.48057E-08 6.21E-07 polypeptide Gadd45gip1 8 84.1 RIKEN cDNA 2310040G17 gene 1417619_at 4 3.38875643 1.4 2 2.69163229 8.84279E-06 0.0001904 BC056474 15 12.1 Mus musculus cDNA clone 1424117_at H3030A06 3.95752801 2.42838452 1.9 2.2 2.62132809 1.3344E-08 5.66E-07 MGC:67360 IMAGE:6823629, complete cds NA 4 153 guanine nucleotide binding protein, 1454696_at -3.46081884 -4 -1.3 -1.6 -2.6026947 8.58458E-05 0.0012617 beta 1 Gnb1 4 153 guanine nucleotide binding protein, 1417432_a_at H3094D02 -3.13334396 -4 -1.6 -1.7 -2.5946297 1.04542E-05 0.0002202 beta 1 Gadd45gip1 8 84.1 RAD23a homolog (S. -

Dual Role of the Mitochondrial Protein Frataxin in Astrocytic Tumors
Laboratory Investigation (2011) 91, 1766–1776 & 2011 USCAP, Inc All rights reserved 0023-6837/11 $32.00 Dual role of the mitochondrial protein frataxin in astrocytic tumors Elmar Kirches1, Nadine Andrae1, Aline Hoefer2, Barbara Kehler1,3, Kim Zarse4, Martin Leverkus3, Gerburg Keilhoff5, Peter Schonfeld5, Thomas Schneider6, Annette Wilisch-Neumann1 and Christian Mawrin1 The mitochondrial protein frataxin (FXN) is known to be involved in mitochondrial iron homeostasis and iron–sulfur cluster biogenesis. It is discussed to modulate function of the electron transport chain and production of reactive oxygen species (ROS). FXN loss in neurons and heart muscle cells causes an autosomal-dominant mitochondrial disorder, Friedreich’s ataxia. Recently, tumor induction after targeted FXN deletion in liver and reversal of the tumorigenic phenotype of colonic carcinoma cells following FXN overexpression were described in the literature, suggesting a tumor suppressor function. We hypothesized that a partial reversal of the malignant phenotype of glioma cells should occur after FXN transfection, if the mitochondrial protein has tumor suppressor functions in these brain tumors. In astrocytic brain tumors and tumor cell lines, we observed reduced FXN levels compared with non-neoplastic astrocytes. Mitochondrial content (citrate synthase activity) was not significantly altered in U87MG glioblastoma cells stably overexpressing FXN (U87-FXN). Surprisingly, U87-FXN cells exhibited increased cytoplasmic ROS levels, although mitochondrial ROS release was attenuated by FXN, as expected. Higher cytoplasmic ROS levels corresponded to reduced activities of glutathione peroxidase and catalase, and lower glutathione content. The defect of antioxidative capacity resulted in increased susceptibility of U87-FXN cells against oxidative stress induced by H2O2 or buthionine sulfoximine. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Genome Editing for the Understanding and Treatment of Inherited Cardiomyopathies
International Journal of Molecular Sciences Review Genome Editing for the Understanding and Treatment of Inherited Cardiomyopathies 1, 1, 1,2, Quynh Nguyen y , Kenji Rowel Q. Lim y and Toshifumi Yokota * 1 Department of Medical Genetics, Faculty of Medicine and Dentistry, University of Alberta, Edmonton, AB T6G2H7, Canada; [email protected] (Q.N.); [email protected] (K.R.Q.L.) 2 The Friends of Garrett Cumming Research & Muscular Dystrophy Canada, HM Toupin Neurological Science Research Chair, Edmonton, AB T6G2H7, Canada * Correspondence: [email protected]; Tel.: +1-780-492-1102 These authors contributed equally to the work. y Received: 28 December 2019; Accepted: 19 January 2020; Published: 22 January 2020 Abstract: Cardiomyopathies are diseases of heart muscle, a significant percentage of which are genetic in origin. Cardiomyopathies can be classified as dilated, hypertrophic, restrictive, arrhythmogenic right ventricular or left ventricular non-compaction, although mixed morphologies are possible. A subset of neuromuscular disorders, notably Duchenne and Becker muscular dystrophies, are also characterized by cardiomyopathy aside from skeletal myopathy. The global burden of cardiomyopathies is certainly high, necessitating further research and novel therapies. Genome editing tools, which include zinc finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs) and clustered regularly interspaced short palindromic repeats (CRISPR) systems have emerged as increasingly important technologies in studying -

The Pathogenetic Role of Β-Cell Mitochondria in Type 2 Diabetes
236 3 Journal of M Fex et al. Mitochondria in β-cells 236:3 R145–R159 Endocrinology REVIEW The pathogenetic role of β-cell mitochondria in type 2 diabetes Malin Fex1, Lisa M Nicholas1, Neelanjan Vishnu1, Anya Medina1, Vladimir V Sharoyko1, David G Nicholls1, Peter Spégel1,2 and Hindrik Mulder1 1Department of Clinical Sciences in Malmö, Unit of Molecular Metabolism, Lund University Diabetes Centre, Clinical Research Center, Malmö University Hospital, Lund University, Malmö, Sweden 2Department of Chemistry, Center for Analysis and Synthesis, Lund University, Sweden Correspondence should be addressed to H Mulder: [email protected] Abstract Mitochondrial metabolism is a major determinant of insulin secretion from pancreatic Key Words β-cells. Type 2 diabetes evolves when β-cells fail to release appropriate amounts f TCA cycle of insulin in response to glucose. This results in hyperglycemia and metabolic f coupling signal dysregulation. Evidence has recently been mounting that mitochondrial dysfunction f oxidative phosphorylation plays an important role in these processes. Monogenic dysfunction of mitochondria is a f mitochondrial transcription rare condition but causes a type 2 diabetes-like syndrome owing to β-cell failure. Here, f genetic variation we describe novel advances in research on mitochondrial dysfunction in the β-cell in type 2 diabetes, with a focus on human studies. Relevant studies in animal and cell models of the disease are described. Transcriptional and translational regulation in mitochondria are particularly emphasized. The role of metabolic enzymes and pathways and their impact on β-cell function in type 2 diabetes pathophysiology are discussed. The role of genetic variation in mitochondrial function leading to type 2 diabetes is highlighted. -

Supplementary File 2A Revised
Supplementary file 2A. Differentially expressed genes in aldosteronomas compared to all other samples, ranked according to statistical significance. Missing values were not allowed in aldosteronomas, but to a maximum of five in the other samples. Acc UGCluster Name Symbol log Fold Change P - Value Adj. P-Value B R99527 Hs.8162 Hypothetical protein MGC39372 MGC39372 2,17 6,3E-09 5,1E-05 10,2 AA398335 Hs.10414 Kelch domain containing 8A KLHDC8A 2,26 1,2E-08 5,1E-05 9,56 AA441933 Hs.519075 Leiomodin 1 (smooth muscle) LMOD1 2,33 1,3E-08 5,1E-05 9,54 AA630120 Hs.78781 Vascular endothelial growth factor B VEGFB 1,24 1,1E-07 2,9E-04 7,59 R07846 Data not found 3,71 1,2E-07 2,9E-04 7,49 W92795 Hs.434386 Hypothetical protein LOC201229 LOC201229 1,55 2,0E-07 4,0E-04 7,03 AA454564 Hs.323396 Family with sequence similarity 54, member B FAM54B 1,25 3,0E-07 5,2E-04 6,65 AA775249 Hs.513633 G protein-coupled receptor 56 GPR56 -1,63 4,3E-07 6,4E-04 6,33 AA012822 Hs.713814 Oxysterol bining protein OSBP 1,35 5,3E-07 7,1E-04 6,14 R45592 Hs.655271 Regulating synaptic membrane exocytosis 2 RIMS2 2,51 5,9E-07 7,1E-04 6,04 AA282936 Hs.240 M-phase phosphoprotein 1 MPHOSPH -1,40 8,1E-07 8,9E-04 5,74 N34945 Hs.234898 Acetyl-Coenzyme A carboxylase beta ACACB 0,87 9,7E-07 9,8E-04 5,58 R07322 Hs.464137 Acyl-Coenzyme A oxidase 1, palmitoyl ACOX1 0,82 1,3E-06 1,2E-03 5,35 R77144 Hs.488835 Transmembrane protein 120A TMEM120A 1,55 1,7E-06 1,4E-03 5,07 H68542 Hs.420009 Transcribed locus 1,07 1,7E-06 1,4E-03 5,06 AA410184 Hs.696454 PBX/knotted 1 homeobox 2 PKNOX2 1,78 2,0E-06 -

Role of Tafazzin in Hematopoiesis and Leukemogenesis
Role of Tafazzin in Hematopoiesis and Leukemogenesis by Ayesh Seneviratne A thesis submitted in conformity with the requirements for the degree of Doctor of Philosophy Institute of Medical Science University of Toronto © Copyright by Ayesh Seneviratne 2020 Role of Tafazzin in Hematopoiesis and Leukemogenesis Ayesh Seneviratne Doctor of Philosophy Institute of Medical Science University of Toronto 2020 Abstract Tafazzin (TAZ) is a mitochondrial transacylase that remodels the mitochondrial cardiolipin into its mature form. Through a CRISPR screen, we identified TAZ as necessary for the growth and viability of acute myeloid leukemia (AML) cells. Genetic inhibition of TAZ reduced stemness and increased differentiation of AML cells both in vitro and in vivo. In contrast, knockdown of TAZ did not impair normal hematopoiesis under basal conditions. Mechanistically, inhibition of TAZ decreased levels of cardiolipin but also altered global levels of intracellular phospholipids, including phosphatidylserine, which controlled AML stemness and differentiation by modulating toll-like receptor (TLR) signaling (Seneviratne et al., 2019). ii Acknowledgments Firstly, I would like to thank Dr. Aaron Schimmer for his guidance and support during my PhD studies. I really enjoyed our early morning meetings where he provided much needed perspective to navigate the road blocks of my project, whilst continuing to push me. It was a privilege to be mentored by such an excellent clinician scientist. I hope to continue to build on the skills I learned in Dr. Schimmer’s lab as I progress on my path to become a clinician scientist. Working in the Schimmer lab was a wonderful learning environment. I would like to especially thank Dr. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Src Inhibitors Modulate Frataxin Protein Levels
Human Molecular Genetics, 2015, Vol. 24, No. 15 4296–4305 doi: 10.1093/hmg/ddv162 Advance Access Publication Date: 6 May 2015 Original Article ORIGINAL ARTICLE Src inhibitors modulate frataxin protein levels Downloaded from https://academic.oup.com/hmg/article/24/15/4296/2453025 by guest on 28 September 2021 Fabio Cherubini1, Dario Serio1, Ilaria Guccini1, Silvia Fortuni1,2, Gaetano Arcuri1, Ivano Condò1, Alessandra Rufini1,2, Shadman Moiz1, Serena Camerini3, Marco Crescenzi3, Roberto Testi1,2 and Florence Malisan1,* 1Laboratory of Signal Transduction, Department of Biomedicine and Prevention, University of Rome ‘Tor Vergata’, Via Montpellier 1, 00133 Rome, Italy, 2Fratagene Therapeutics Ltd, 22 Northumberland Rd, Dublin, Ireland and 3Department of Cell Biology and Neurosciences, Italian National Institute of Health, Viale Regina Elena, 299, 00161 Rome, Italy *To whom correspondence should be addressed at: Laboratory of Signal Transduction, Department of Biomedicine and Prevention, University of Rome ‘Tor Vergata’, Via Montpellier 1, 00133 Rome, Italy. Tel: +39 0672596501; Fax: +39 0672596505; Email: [email protected] Abstract Defective expression of frataxin is responsible for the inherited, progressive degenerative disease Friedreich’s Ataxia (FRDA). There is currently no effective approved treatment for FRDA and patients die prematurely. Defective frataxin expression causes critical metabolic changes, including redox imbalance and ATP deficiency. As these alterations are known to regulate the tyrosine kinase Src, we investigated whether Src might in turn affect frataxin expression. We found that frataxin can be phosphorylated by Src. Phosphorylation occurs primarily on Y118 and promotes frataxin ubiquitination, a signal for degradation. Accordingly, Src inhibitors induce accumulation of frataxin but are ineffective on a non-phosphorylatable frataxin- Y118F mutant. -
Grantsummaries 2014-2017 Jf
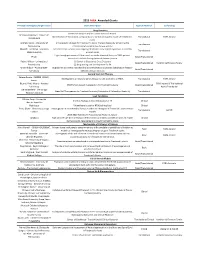
2015 FARA Awarded Grants Principal Investigator/Organization Grant Description Type of Research Co-funding Drug Discovery 2014 Keith Michael Andrus Cardiac Research Award: Veronique Monnier - Université Identification of therapeutic compounds on a cardiac Drosophila model of Friedreich’s Translational FARA Ireland Paris Diderot ataxia Andrew Dancis - University of A therapeutic strategy for Friedreich's ataxia: Frataxin bypass by enhancing the Translational Pennsylvania mitochondrial cysteine desulfurase activity Edward L. Grabczyk - Louisiana Small molecule induced exon skipping of MLH3 to slow repeat expansion in an FRDA Translational State University mousel model High throughput screen of Pfizer small molecule chemical library in FRDA patient- Pfizer Basic/Translational derived cells to look for regulators of frataxin protein Robert Wilson - University of (1) Center of Excellence: Drug Discovery Basic/Translational Hamilton & Finneran Family Pennsylvania (2) Drug testing and development for FA Omar Khdour - Arizona State Approaches to correct mitochondrial abnormalities and synaptic pathology in frataxin- Basic/Translational University deficient sensory neurons Gene & Stem Cell Therapy Helene Puccio - INSERM, IGBMC, Development of neuronal gene therapy for the treatment of FRDA Translational FARA Ireland France Ricardo Pinto Mouro - Harvard FARA Ireland & The National CRISPR/Cas9-based Therapeutics for Friedreich's ataxia Basic/Translational University Ataxia Foundation Joel Gottesfeld - The Scripps Stem Cell Therapeutics for Friedreich's