A Novel Rhabdovirus Infecting Newly Discovered Nycteribiid Bat Flies
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Entwicklung Einer Software Zur Identifizierung Neuartiger Und
Entwicklung einer Software zur Identifizierung neuartiger und bekannter Infektionserreger in klinischen Proben Dissertation zur Erlangung des Doktorgrades an der Fakult¨at fur¨ Mathematik, Informatik und Naturwissenschaften Fachbereich Biologie der Universit¨at Hamburg vorgelegt von Malik Alawi Hamburg, 2020 Vorsitzender der Prufungskommission¨ Dr. PD Andreas Pommerening-R¨oser Gutachter Professor Dr. Adam Grundhoff Professor Dr. Stefan Kurtz Datum der Disputation 30. April 2021 Abstract Sequencing of diagnostic samples is widely considered a key technology that may fun- damentally improve infectious disease diagnostics. The approach can not only identify pathogens already known to cause a specific disease, but may also detect pathogens that have not been previously attributed to this disease, as well as completely new, previously unknown pathogens. Therefore, it may significantly increase the level of preparedness for future outbreaks of emerging pathogens. This study describes the development and application of methods for the identification of pathogenic agents in diagnostic samples. The methods have been successfully applied multiple times under clinical conditions. The corresponding results have been published within the scope of this thesis. Finally, the methods were made available to the scientific community as an open source bioinformatics tool. The novel software was validated by conventional diagnostic methods and it was compared to established analysis pipelines using authentic clinical samples. It is able to identify pathogens from different diagnostic entities and often classifies viral agents down to strain level. Furthermore, the method is capable of assembling complete viral genomes, even from samples containing multiple closely related viral strains of the same viral family. In addition to an improved method for taxonomic classification, the software offers functionality which is not present in established analysis pipelines. -

Using Cell Lines to Study Factors Affecting Transmission of Fish Viruses
Using cell lines to study factors affecting transmission of fish viruses by Phuc Hoang Pham A thesis presented to the University of Waterloo in fulfillment of the thesis requirement for the degree of Doctor of Philosophy in Biology Waterloo, Ontario, Canada, 2014 ©Phuc Hoang Pham 2014 AUTHOR'S DECLARATION I hereby declare that I am the sole author of this thesis. This is a true copy of the thesis, including any required final revisions, as accepted by my examiners. I understand that my thesis may be made electronically available to the public. ii ABSTRACT Factors that can influence the transmission of aquatic viruses in fish production facilities and natural environment are the immune defense of host species, the ability of viruses to infect host cells, and the environmental persistence of viruses. In this thesis, fish cell lines were used to study different aspects of these factors. Five viruses were used in this study: viral hemorrhagic septicemia virus (VHSV) from the Rhabdoviridae family; chum salmon reovirus (CSV) from the Reoviridae family; infectious pancreatic necrosis virus (IPNV) from the Birnaviridae family; and grouper iridovirus (GIV) and frog virus-3 (FV3) from the Iridoviridae family. The first factor affecting the transmission of fish viruses examined in this thesis is the immune defense of host species. In this work, infections of marine VHSV-IVa and freshwater VHSV-IVb were studied in two rainbow trout cell lines, RTgill-W1 from the gill epithelium, and RTS11 from spleen macrophages. RTgill-W1 produced infectious progeny of both VHSV-IVa and -IVb. However, VHSV-IVa was more infectious than IVb toward RTgill-W1: IVa caused cytopathic effects (CPE) at a lower viral titre, elicited CPE earlier, and yielded higher titres. -

An Open Source Bioinformatics Tool for Fast, Systematic and Cohort Based
www.nature.com/scientificreports OPEN DAMIAN: an open source bioinformatics tool for fast, systematic and cohort based analysis of microorganisms in diagnostic samples Malik Alawi 1,2, Lia Burkhardt1, Daniela Indenbirken1, Kerstin Reumann1, Maximilian Christopeit3, Nicolaus Kröger3, Marc Lütgehetmann4, Martin Aepfelbacher4, Nicole Fischer4,5* & Adam Grundhof 1,5* We describe DAMIAN, an open source bioinformatics tool designed for the identifcation of pathogenic microorganisms in diagnostic samples. By using authentic clinical samples and comparing our results to those from established analysis pipelines as well as conventional diagnostics, we demonstrate that DAMIAN rapidly identifes pathogens in diferent diagnostic entities, and accurately classifes viral agents down to the strain level. We furthermore show that DAMIAN is able to assemble full-length viral genomes even in samples co-infected with multiple virus strains, an ability which is of considerable advantage for the investigation of outbreak scenarios. While DAMIAN, similar to other pipelines, analyzes single samples to perform classifcation of sequences according to their likely taxonomic origin, it also includes a tool for cohort-based analysis. This tool uses cross-sample comparisons to identify sequence signatures that are frequently present in a sample group of interest (e.g., a disease- associated cohort), but occur less frequently in control cohorts. As this approach does not require homology searches in databases, it principally allows the identifcation of not only known, but also completely novel pathogens. Using samples from a meningitis outbreak, we demonstrate the feasibility of this approach in identifying enterovirus as the causative agent. Nucleic acid based detection of pathogens has widely replaced culture based laboratory methods for the identif- cation of putative pathogens in samples from patients with infectious diseases1,2. -

Bat Rabies Surveillance in Europe J
Zoonoses and Public Health SPECIALISSUE–BATS Bat Rabies Surveillance in Europe J. Schatz1, A. R. Fooks2, L. McElhinney2, D. Horton2, J. Echevarria3,S.Va´ zquez-Moron3, E. A. Kooi4, T. B. Rasmussen5,T.Mu¨ ller1 and C. M. Freuling1 1 Institute of Molecular Biology, WHO Collaborating Centre for Rabies Surveillance and Research, Friedrich-Loeffler-Institute, Federal Research Institute for Animal Health, Greifswald-Insel Riems, Germany 2 Wildlife Zoonoses and Vector Borne Diseases Research Group, Animal Health and Veterinary Laboratories Agency (Weybridge), Surrey, UK 3 Instituto de Salud Carlos III, Majadahonda, Madrid, Spain 4 Central Veterinary Institute of Wageningen UR, Lelystad, The Netherlands 5 DTU National Veterinary Institute, Lindholm, Kalvehave, Denmark Impacts • Rabies in European bats is caused by at least four different lyssavirus species that seem to have an association with certain bat species, that is, the Serotine bat (EBLV-1), the Daubenton’s and Pond bat (EBLV-2), Schreiber’s long-fingered bat (WCBV) and the Natterer’s bat (BBLV). • Almost all bat rabies cases were detected in dead or clinically affected bats i.e. passive surveillance. This is a much more sensitive approach than active surveillance where oral swab samples taken from individuals randomly captured during planned surveys were screened for viral RNA and/or virus. • The focus of bat rabies surveillance should be on testing of dead or moribund animals, and resulting positive FAT results should be further analysed to identify the lyssavirus species and all bats submitted for testing should be identified to species level. Keywords: Summary Rabies; bats; lyssavirus; surveillance Rabies is the oldest known zoonotic disease and was also the first recognized bat Correspondence: associated infection in humans. -

For Review Only Journal of Zoological Systematics and Evolutionary Research Page 2 of 47
Journal of Zoological Systematics and Evolutionary Research Comparative phylogeography and asymmetric hybridization between cryptic bat species Journal: Journal of Zoological Systematics and Evolutionary Research Manuscript ID JZS.201900005.R2 Wiley - Manuscript type:ForOriginal Review Article Only Date Submitted by the n/a Author: Complete List of Authors: Centeno-Cuadros, Alejandro; Universidad de Cádiz Facultad de Ciencias del Mar y Ambientales, Biomedicina, Biotecnología y Salud Pública Razgour, Orly; University of Southampton Centre for Biological Sciences, Centre for Biological Sciences García-Mudarra, Juan Luís; Estacion Biologica de Donana CSIC, Ecología Evolutiva Mingo-Casas, Patricia; Instituto de Salud Carlos III Campus de Majadahonda, Instituto de Salud Carlos III Sandonís, Virginia; Instituto de Salud Carlos III Campus de Majadahonda, Instituto de Salud Carlos III Redondo, Adrián; Estacion Biologica de Donana CSIC, Ecología Evolutiva Ibáñez, Carlos; Estacion Biologica de Donana CSIC, Ecología Evolutiva de Paz, Óscar; Universidad de Alcala de Henares, Ciencias de la Vida Martinez-Alós, Susana; Universidad de Alcala de Henares, Ciencias de la Vida Pérez Suarez, Gonzalo; Universidad de Alcala de Henares, Ciencias de la Vida Echevarría, Juan; Instituto de Salud Carlos III Campus de Majadahonda, Instituto de Salud Carlos III; CIBER de Epidemiología y Salud Pública, CIBERESP Juste, Javier; Estacion Biologica de Donana CSIC, Ecología Evolutiva Eptesicus, isabellinus, serotinus, Approximate Bayesian Computation, Keywords: hybrids Cryptic speciation and hybridization are two key processes that affect the origin and maintenance of biodiversity and our ability to understand and estimate it. To determine how these two processes interact, we studied allopatric and sympatric colonies of two cryptic bat species (Eptesicus serotinus and E. isabellinus) with parapatric distribution in the Iberian Peninsula. -

(Townsend's Big-Eared Bat), Eptesicus Fuscus
América do Norte Antrozous pallidus (Pallid), Corynorhinus townsendii (Townsend's big-eared bat), Eptesicus fuscus (Big brown), Euderma maculatum (Spotted), Eumops floridanus (Bonneted), Eumops perotis (Western mastiff), Eumops underwoodi (Underwood's bonneted), Lasionycteris noctivagans (Silver- haired), Lasiurus blossevilli (Western red), Lasiurus borealis (Eastern red), Lasiurus cinereus (Hoary), Lasiurus ega (Southern yellow), Lasiurus intermedius (Northern yellow), Lasiurus seminolus (S eminole bat), Lasiurus xanthinus (Western yellow), Macrotus californicus (California leaf-nosed), Molossus molossus (Pallas's mastiff), Mormoops megalophylla (Ghost faced), Myotis austroriparius (Southeastern myotis), Myotis californicus (California myotis), Myotis ciliolabrum (Western Small-footed), Myotis evotis (Long- eared myotis), Myotis grisescens (Gray), Myotis leibii (Small-footed), Myotis lucifugus (Little brown), Myotis occultus (Arizona myotis), Myotis septentrionalis (Northern long-eared myotis), Myotis sodalis (Indiana), Myotis thysanodes (Fringed myotis), Myotis velifer (Cave myotis), Myotis volans (Long-legged myotis), Myotis yumanensis (Yuma myotis), Nycticeius humeralis (Evening), Nyctinomops femorosaccus (Pocketed free-tailed), Nyctinomops macrotis (Big free-tailed), Parastrellus hesperus (Western pipistrelle), Perimyotis subflavus (Tricolored), Tadarida brasiliensis (Mexican free-tailed) Reino unido e Europa Barbastella barbastellus (Western barbastelle)*, Eptesicus isabellinus (Meridional serotine), Eptesicus isabellinus (Meridional -

Zoologische Mededelingen
ZOOLOGISCHE MEDEDELINGEN UITGEGEVEN DOOR HET RIJKSMUSEUM VAN NATUURLIJKE HISTORIE TE LEIDEN (MINISTERIE VAN CULTUUR, RECREATIE EN MAATSCHAPPELIJK WERK) Deel 55 no. 14 4 maart 1980 A NEW FRUIT BAT OF THE GENUS MYONYCTERIS MATSCHIE, 1899, FROM EASTERN KENYA AND TANZANIA (MAMMALIA, MEGACHIROPTERA) by W. BERGMANS Instituut voor Taxonomische Zoölogie, Universiteit van Amsterdam With 4 text-figures ABSTRACT Myonycteris relicta n. sp. is described from the Shimba Hills in southeast Kenya and from the Usambara Mountains in northeast Tanzania. The species is larger than the only other known African mainland species of the genus, Myonycteris torquata (Dobson, 1878), from the Central and West African rain forests and, if compared to M. torquata and the only other species in the genus, M. brachycephala (Bocage, 1889) from São Tomé, has a relatively longer rostrum, a more deflected cranial axis, and further differs in number, shape and position of its teeth. The new species provides new arguments for the relationship between the genera Myonycteris Matschie, 1899, and Lissonycteris Andersen, 1912. It is believed that Myonycteris relicta may be a forest species and as such restricted to isolated East African forests. INTRODUCTION During a visit to the Zoologisches Museum in Berlin (ZMB), in April 1979, the author found two fruit bat specimens from the Tanzanian Usa- mbara Mountains, which proved to represent an undescribed taxon. Later, in June 1979, Dr C. Smeenk of the Rijksmuseum van Natuurlijke Historie at Leiden (RMNH) recognized a third specimen of this taxon in newly acquired material from the Shimba Hills in southeast Kenya. The bats differ on specific level from all other known fruit bats, and are described in the present paper. -
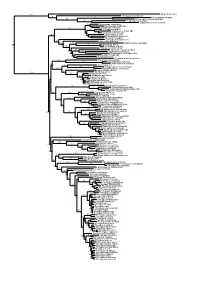
Figs1 ML Tree.Pdf
100 Megaderma lyra Rhinopoma hardwickei 71 100 Rhinolophus creaghi 100 Rhinolophus ferrumequinum 100 Hipposideros armiger Hipposideros commersoni 99 Megaerops ecaudatus 85 Megaerops niphanae 100 Megaerops kusnotoi 100 Cynopterus sphinx 98 Cynopterus horsfieldii 69 Cynopterus brachyotis 94 50 Ptenochirus minor 86 Ptenochirus wetmorei Ptenochirus jagori Dyacopterus spadiceus 99 Sphaerias blanfordi 99 97 Balionycteris maculata 100 Aethalops alecto 99 Aethalops aequalis Thoopterus nigrescens 97 Alionycteris paucidentata 33 99 Haplonycteris fischeri 29 Otopteropus cartilagonodus Latidens salimalii 43 88 Penthetor lucasi Chironax melanocephalus 90 Syconycteris australis 100 Macroglossus minimus 34 Macroglossus sobrinus 92 Boneia bidens 100 Harpyionycteris whiteheadi 69 Harpyionycteris celebensis Aproteles bulmerae 51 Dobsonia minor 100 100 80 Dobsonia inermis Dobsonia praedatrix 99 96 14 Dobsonia viridis Dobsonia peronii 47 Dobsonia pannietensis 56 Dobsonia moluccensis 29 Dobsonia anderseni 100 Scotonycteris zenkeri 100 Casinycteris ophiodon 87 Casinycteris campomaanensis Casinycteris argynnis 99 100 Eonycteris spelaea 100 Eonycteris major Eonycteris robusta 100 100 Rousettus amplexicaudatus 94 Rousettus spinalatus 99 Rousettus leschenaultii 100 Rousettus aegyptiacus 77 Rousettus madagascariensis 87 Rousettus obliviosus Stenonycteris lanosus 100 Megaloglossus woermanni 100 91 Megaloglossus azagnyi 22 Myonycteris angolensis 100 87 Myonycteris torquata 61 Myonycteris brachycephala 33 41 Myonycteris leptodon Myonycteris relicta 68 Plerotes anchietae -

Comparison of Plant‐Adapted Rhabdovirus Protein Localization and Interactions
University of Kentucky UKnowledge University of Kentucky Doctoral Dissertations Graduate School 2011 COMPARISON OF PLANT‐ADAPTED RHABDOVIRUS PROTEIN LOCALIZATION AND INTERACTIONS Kathleen Marie Martin University of Kentucky, [email protected] Right click to open a feedback form in a new tab to let us know how this document benefits ou.y Recommended Citation Martin, Kathleen Marie, "COMPARISON OF PLANT‐ADAPTED RHABDOVIRUS PROTEIN LOCALIZATION AND INTERACTIONS" (2011). University of Kentucky Doctoral Dissertations. 172. https://uknowledge.uky.edu/gradschool_diss/172 This Dissertation is brought to you for free and open access by the Graduate School at UKnowledge. It has been accepted for inclusion in University of Kentucky Doctoral Dissertations by an authorized administrator of UKnowledge. For more information, please contact [email protected]. ABSTRACT OF DISSERTATION Kathleen Marie Martin The Graduate School University of Kentucky 2011 COMPARISON OF PLANT‐ADAPTED RHABDOVIRUS PROTEIN LOCALIZATION AND INTERACTIONS ABSTRACT OF DISSERTATION A dissertation submitted in partial fulfillment of the requirements for the Degree of Doctor of Philosophy in the College of Agriculture at the University of Kentucky By Kathleen Marie Martin Lexington, Kentucky Director: Dr. Michael M Goodin, Associate Professor of Plant Pathology Lexington, Kentucky 2011 Copyright © Kathleen Marie Martin 2011 ABSTRACT OF DISSERTATION COMPARISON OF PLANT‐ADAPTED RHABDOVIRUS PROTEIN LOCALIZATION AND INTERACTIONS Sonchus yellow net virus (SYNV), Potato yellow dwarf virus (PYDV) and Lettuce Necrotic yellows virus (LNYV) are members of the Rhabdoviridae family that infect plants. SYNV and PYDV are Nucleorhabdoviruses that replicate in the nuclei of infected cells and LNYV is a Cytorhabdovirus that replicates in the cytoplasm. LNYV and SYNV share a similar genome organization with a gene order of Nucleoprotein (N), Phosphoprotein (P), putative movement protein (Mv), Matrix protein (M), Glycoprotein (G) and Polymerase protein (L). -

4 Introduction
4 Introduction In 1967, in Marburg an der Lahn and Frankfurt ‘subtypes’ of a novel agent named ‘Ebola virus’ am Main, Germany1, and in Belgrade, Yugoslavia after the small Ebola river in Zaire [412, 1000, (now Serbia), laboratory workers accepted shipments 2410]. Today these ‘subtypes’ are called Sudan of African green monkeys (Chlorocebus aethiops) ebolavirus (SEBOV) and Zaire ebolavirus (ZEBOV), from Uganda. As they had done many times before respectively [805]. Studies of periodic hemorrhagic with such animals, workers performed routine ex- fever outbreaks in African countries and in the aminations for apparent ailments and then prepared Philippines indicated that at least two more ebola- tissue cultures from the monkeys’ kidneys for the viruses exist, which are now known as the Coote^ development of poliomyelitis vaccines. A few days d’Ivoire ebolavirus (CIEBOV) and Reston ebola- later, several workers were reported ill and were virus (REBOV) [805]. Molecular and other studies admitted to local hospitals. A total of 32 people revealed the close relationship of MARV and the fell sick with an apparently new disease, of which ebolaviruses, which resulted in their classification seven died. A hitherto unknown virus was isolated in the same viral family, Filoviridae (the filo- from patients and human tissues [2396] and called viruses2) [805]. ‘Marburg virus’ (today Lake Victoria marburgvirus, A substantial interest in filoviruses has developed MARV) [805]. Over the subsequent three decades, among the general public, in part because of novels, only individual MARV infections were recorded. popular science stories, and Hollywood productions In 1998, the virus reappeared in the Democratic that portrayed the horrendous diseases they cause. -

Complete Sections As Applicable
This form should be used for all taxonomic proposals. Please complete all those modules that are applicable (and then delete the unwanted sections). For guidance, see the notes written in blue and the separate document “Help with completing a taxonomic proposal” Please try to keep related proposals within a single document; you can copy the modules to create more than one genus within a new family, for example. MODULE 1: TITLE, AUTHORS, etc (to be completed by ICTV Code assigned: 2016.017aM officers) Short title: One (1) new species in the genus Cytorhabdovirus, family Rhabdoviridae (e.g. 6 new species in the genus Zetavirus) Modules attached 2 3 4 5 (modules 1 and 11 are required) 6 7 8 9 10 Author(s): Colleen M. Higgins, Nicolas Bejerman, Ming Li, Anthony P. James, Ralf G. Dietzgen, Michael N. Pearson, Peter A. Revill, Robert M. Harding Corresponding author with e-mail address: Colleen Higgins; [email protected] List the ICTV study group(s) that have seen this proposal: A list of study groups and contacts is provided at http://www.ictvonline.org/subcommittees.asp . If ICTV Rhabdoviridae Study Group in doubt, contact the appropriate subcommittee chair (fungal, invertebrate, plant, prokaryote or vertebrate viruses) ICTV Study Group comments (if any) and response of the proposer: 11 members have advised support for the proposal; 1 member has not responded. Date first submitted to ICTV: 18 July, 2016 Date of this revision (if different to above): ICTV-EC comments and response of the proposer: Page 1 of 7 MODULE 2: NEW SPECIES creating and naming one or more new species. -

Final Report on the Project
BP Conservation Programme (CLP) 2005 PROJECT NO. 101405 - BRONZE AWARD WINNER ECOLOGY, DISTRIBUTION, STATUS AND PROTECTION OF THREE CONGOLESE FRUIT BATS FINAL REPORT Patrick KIPALU Team Leader Observatoire Congolais pour la Protection de l’Environnement OCPE – ong Kinshasa – Democratic Republic of the Congo E-mail: [email protected] APRIL 2009 1 Table of Content Acknowledgements…………………………………………………………………. p3 I. Project Summary……………………………………………………………….. p4 II. Introduction…………………………………………………………………… p4-p7 III. Materials and Methods ……………………………………………………….. p7-p10 IV. Results per Study Site…………………………………………………………. p10-p15 1. Pointe-Noire ………………………………………………………….. p10-p12 2. Mayumbe Forest /Luki Reserve……………………………………….. p12-p13 3. Zongo Forest…………………………………………………………... p14 4. Mbanza-Ngungu ………………………………………………………. P15 V. General Results ………………………………………………………………p15-p16 VI. Discussions……………………………………………………………………p17-18 VII. Conclusion and Recommendations……………………………………….p 18-p19 VIII. Bibliography………………………………………………………………p20-p21 Acknowledgements 2 The OCPE (Observatoire Congolais pour la Protection de l’Environnement) project team would like to start by expressing our gratefulness and saying thank you to the BP Conservation Program, which has funded the execution of this project. The OCPE also thanks the Van Tienhoven Foundation which provided a further financial support. Without these organisations, execution of the project would not have been possible. We would like to thank specially the BPCP “dream team”: Marianne D. Carter, Robyn Dalzen and our regretted Kate Stoke for their time, advices, expertise and care, which helped us to complete this work, Our special gratitude goes to Dr. Wim Bergmans, who was the hero behind the scene from the conception to the execution of the research work. Without his expertise, advices and network it would had been difficult for the project team to produce any result from this project.