Paul Mcbride a Thesis Submitted to Auckland University of Technology
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Amphibious Fishes: Terrestrial Locomotion, Performance, Orientation, and Behaviors from an Applied Perspective by Noah R
AMPHIBIOUS FISHES: TERRESTRIAL LOCOMOTION, PERFORMANCE, ORIENTATION, AND BEHAVIORS FROM AN APPLIED PERSPECTIVE BY NOAH R. BRESSMAN A Dissertation Submitted to the Graduate Faculty of WAKE FOREST UNIVESITY GRADUATE SCHOOL OF ARTS AND SCIENCES in Partial Fulfillment of the Requirements for the Degree of DOCTOR OF PHILOSOPHY Biology May 2020 Winston-Salem, North Carolina Approved By: Miriam A. Ashley-Ross, Ph.D., Advisor Alice C. Gibb, Ph.D., Chair T. Michael Anderson, Ph.D. Bill Conner, Ph.D. Glen Mars, Ph.D. ACKNOWLEDGEMENTS I would like to thank my adviser Dr. Miriam Ashley-Ross for mentoring me and providing all of her support throughout my doctoral program. I would also like to thank the rest of my committee – Drs. T. Michael Anderson, Glen Marrs, Alice Gibb, and Bill Conner – for teaching me new skills and supporting me along the way. My dissertation research would not have been possible without the help of my collaborators, Drs. Jeff Hill, Joe Love, and Ben Perlman. Additionally, I am very appreciative of the many undergraduate and high school students who helped me collect and analyze data – Mark Simms, Tyler King, Caroline Horne, John Crumpler, John S. Gallen, Emily Lovern, Samir Lalani, Rob Sheppard, Cal Morrison, Imoh Udoh, Harrison McCamy, Laura Miron, and Amaya Pitts. I would like to thank my fellow graduate student labmates – Francesca Giammona, Dan O’Donnell, MC Regan, and Christine Vega – for their support and helping me flesh out ideas. I am appreciative of Dr. Ryan Earley, Dr. Bruce Turner, Allison Durland Donahou, Mary Groves, Tim Groves, Maryland Department of Natural Resources, UF Tropical Aquaculture Lab for providing fish, animal care, and lab space throughout my doctoral research. -

The Morphology and Evolution of Tooth Replacement in the Combtooth Blennies
The morphology and evolution of tooth replacement in the combtooth blennies (Ovalentaria: Blenniidae) A THESIS SUBMITTED TO THE FACULTY OF THE UNIVERSITY OF MINNESOTA BY Keiffer Logan Williams IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF MASTER OF SCIENCE Andrew M. Simons July 2020 ©Keiffer Logan Williams 2020 i ACKNOWLEDGEMENTS I thank my adviser, Andrew Simons, for mentoring me as a student in his lab. His mentorship, kindness, and thoughtful feedback/advice on my writing and research ideas have pushed me to become a more organized and disciplined thinker. I also like to thank my committee: Sharon Jansa, David Fox, and Kory Evans for feedback on my thesis and during committee meetings. An additional thank you to Kory, for taking me under his wing on the #backdattwrasseup project. Thanks to current and past members of the Simons lab/office space: Josh Egan, Sean Keogh, Tyler Imfeld, and Peter Hundt. I’ve enjoyed the thoughtful discussions, feedback on my writing, and happy hours over the past several years. Thanks also to the undergraduate workers in the Simons lab who assisted with various aspects of my work: Andrew Ching and Edward Hicks for helping with histology, and Alex Franzen and Claire Rude for making my terms as curatorial assistant all the easier. In addition, thank you to Kate Bemis and Karly Cohen for conducting a workshop on histology to collect data for this research, and for thoughtful conversations and ideas relating to this thesis. Thanks also to the University of Guam and Laurie Raymundo for hosting me as a student to conduct fieldwork for this research. -

Supplemental Wing Shape and Dispersal Analysis
Data Supplement High dispersal ability inhibits speciation in a continental radiation of passerine birds Santiago Claramunt, Elizabeth P. Derryberry, J. V. Remsen, Jr. & Robb T. Brumfield Museum of Natural Science and Department of Biological Sciences, Louisiana State University, Baton Rouge, LA 70803, USA HAND-WING INDEX AND FLIGHT PERFORMANCE IN NEOTROPICAL FOREST BIRDS We investigated the relationship between wing shape and flight distances determined during 'dispersal challenge' experiments conducted in Gatun Lake in the Panama Canal (Moore et al. 2008). During the experiments, birds were released from a boat at incremental distances from shore and the distance flown or the success or failure in reaching the coast was recorded. To investigated the relationship between the hand-wing index and flight distance in Neotropical birds we used data on mean distance flown from table 3 in ref. We estimated hand-wing indices for the 10 species reported in those experiments (Table S1) . Wing measurements were taken by SC for four males of each species at LSUMNS. The relationship between the hand-wing index and distance flown was evaluated statistically using phylogenetic generalized least-squares (PGLS, Freckleton et al. 2002). We generated a phylogeny for the species involved in the experiment or an appropriate surrogate using DNA sequences of the slow-evolving RAG 1 gene from GenBank (Table S2). A maximum likelihood ultrametric tree was generated in PAUP* (Swofford 2003) using a GTR+! model of nucleotide substitution rates, empirical nucleotide frequencies, and enforcing a molecular clock. We found that the hand-wing index was strongly related to mean distance flown (R2 = 0.68, F = 20, d.f. -

Phylogenetic Analysis of the Nest Architecture of Neotropical Ovenbirds (Furnariidae)
The Auk 116(4):891-911, 1999 PHYLOGENETIC ANALYSIS OF THE NEST ARCHITECTURE OF NEOTROPICAL OVENBIRDS (FURNARIIDAE) KRZYSZTOF ZYSKOWSKI • AND RICHARD O. PRUM NaturalHistory Museum and Department of Ecologyand Evolutionary Biology, University of Kansas,Lawrence, Kansas66045, USA ABSTRACT.--Wereviewed the tremendousarchitectural diversity of ovenbird(Furnari- idae) nestsbased on literature,museum collections, and new field observations.With few exceptions,furnariids exhibited low intraspecificvariation for the nestcharacters hypothe- sized,with the majorityof variationbeing hierarchicallydistributed among taxa. We hy- pothesizednest homologies for 168species in 41 genera(ca. 70% of all speciesand genera) and codedthem as 24 derivedcharacters. Forty-eight most-parsimonious trees (41 steps,CI = 0.98, RC = 0.97) resultedfrom a parsimonyanalysis of the equallyweighted characters using PAUP,with the Dendrocolaptidaeand Formicarioideaas successiveoutgroups. The strict-consensustopology based on thesetrees contained 15 cladesrepresenting both tra- ditionaltaxa and novelphylogenetic groupings. Comparisons with the outgroupsdemon- stratethat cavitynesting is plesiomorphicto the furnariids.In the two lineageswhere the primitivecavity nest has been lost, novel nest structures have evolved to enclosethe nest contents:the clayoven of Furnariusand the domedvegetative nest of the synallaxineclade. Althoughour phylogenetichypothesis should be consideredas a heuristicprediction to be testedsubsequently by additionalcharacter evidence, this first cladisticanalysis -
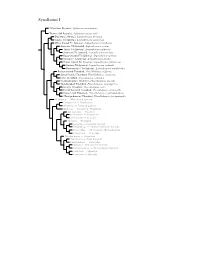
Synallaxini Species Tree
Synallaxini I ?Masafuera Rayadito, Aphrastura masafuerae Thorn-tailed Rayadito, Aphrastura spinicauda Des Murs’s Wiretail, Leptasthenura desmurii Tawny Tit-Spinetail, Leptasthenura yanacensis White-browed Tit-Spinetail, Leptasthenura xenothorax Araucaria Tit-Spinetail, Leptasthenura setaria Tufted Tit-Spinetail, Leptasthenura platensis Striolated Tit-Spinetail, Leptasthenura striolata Rusty-crowned Tit-Spinetail, Leptasthenura pileata Streaked Tit-Spinetail, Leptasthenura striata Brown-capped Tit-Spinetail, Leptasthenura fuliginiceps Andean Tit-Spinetail, Leptasthenura andicola Plain-mantled Tit-Spinetail, Leptasthenura aegithaloides Rufous-fronted Thornbird, Phacellodomus rufifrons Streak-fronted Thornbird, Phacellodomus striaticeps Little Thornbird, Phacellodomus sibilatrix Chestnut-backed Thornbird, Phacellodomus dorsalis Spot-breasted Thornbird, Phacellodomus maculipectus Greater Thornbird, Phacellodomus ruber Freckle-breasted Thornbird, Phacellodomus striaticollis Orange-eyed Thornbird, Phacellodomus erythrophthalmus ?Orange-breasted Thornbird, Phacellodomus ferrugineigula Hellmayrea — White-tailed Spinetail Coryphistera — Brushrunner Anumbius — Firewood-gatherer Asthenes — Canasteros, Thistletails Acrobatornis — Graveteiro Metopothrix — Plushcrown Xenerpestes — Graytails Siptornis — Prickletail Roraimia — Roraiman Barbtail Thripophaga — Speckled Spinetail, Softtails Limnoctites — ST Spinetail, SB Reedhaunter Cranioleuca — Spinetails Pseudasthenes — Canasteros Spartonoica — Wren-Spinetail Pseudoseisura — Cacholotes Mazaria – White-bellied -

Splits, Lumps and Shuffles Splits, Lumps and Shuffles Alexander C
>> SPLITS, LUMPS AND SHUFFLES Splits, lumps and shuffles Alexander C. Lees This series focuses on recent taxonomic proposals—be they entirely new species, splits, lumps or reorganisations—that are likely to be of greatest interest to birders. This latest instalment includes papers relating to a stunning new species of storm petrel, a barrage of rail, woodpecker and tyrannulet splits, insights into some duck, parrot and parrotlet relationships, the to-be-expected furnariid splits, lumps and shuffles (note however not an antbird or tapaculo in sight) and more analyses of old favourites such a Common Bush Tanagers and Rufous-naped Wrens. Get your lists out! A new storm petrel from Chile cyanoptera occurring from southern Peru and southern Brazil to Tierra del Fuego. Wilson et The saga of the uniquely-patterned storm petrels al. (2013) investigated patterns of genetic and first seen on ferry crossings in the Puerto Montt phenotypic divergence between the small-bodied and Chacao channel area (crossing to the Chiloé lowland A. c. cyanoptera and the larger bodied Archipelago), Chile, has finally ended with the highland A. c. orinomus which inhabits hypoxic formal description of a new species. First seen in (low oxygen) Andean water bodies. The subspecies the field as long ago as 1983, it wasn’t until the orinomus is significantly larger with significant publication of a series of images (O’Keeffe et al. frequency differences in a single α-hemoglobin 2009) that the wider birding public became aware amino acid polymorphism (adaptions both to of this undescribed taxon. However, all-the-while, the cold and to low oxygen environments). -

0555 NIA II, 551 AB, Monday 12 July 2010 Carlos Donascimiento1
0555 NIA II, 551 AB, Monday 12 July 2010 Carlos DoNascimiento1, John Lundberg2, Mark Sabaj Pérez2, Nadia Milani3 1Universidad de Carabobo, Valencia, Carabobo, Venezuela, 2The Academy of Natural Sciences, Philadelphia, PA, United States, 3Universidad Central de Venezuela, Caracas, Distrito Capital, Venezuela An Unexpectedly Diverse Group of Miniature and Sexually Dimorphic Neotropical Catfishes Representing a New Genus (Siluriformes, Heptapteridae) Small body size has been a main limiting factor veiling our knowledge of the Neotropical fish diversity, with most of the currently known miniature species described in the last few decades. Frequently, these are found in museum collections catalogued as immature stages, this being the case for the species reported here. Independent collecting efforts in the Peruvian Amazon and Venezuelan Orinoco as well as a revision of material already available in museums has resulted in the recognition of at least five different species of tiny catfishes, that were identified either as heptapterid juveniles or in the slightly more accurate cases as Imparfinis juveniles. Nonetheless, a detailed morphological study revealed that they represent fully mature individuals, easily assignable to the Nemuroglanis subclade of Heptapteridae, but not to Imparfinis or to any other available name in that family. Morphology of the pectoral girdle and fin exhibits striking contrasting conditions between males and females, and along with modifications of the most anterior ribs, also indicate that they constitute a monophyletic group that is here proposed as a new genus. Derived traits of the transverse process of the fourth vertebra, postcleithral process and head laterosensory system support a sister group relationship between Horiomyzon and this new genus, indicating that a single miniaturization event occurred for this subgroup of heptapterids. -

Life History Changes with the Colonisation of Land by Fish
Life history changes with the colonisation of land by fish Edward Richard Murray Platt Supervised by: Terry Ord THESIS SUBMITTED FOR THE DEGREE OF MASTER OF PHILOSOPHY Evolution and Ecology Research Centre School of Biological, Earth and Environmental Sciences Faculty of Science University of New South Wales March 2014 THE UNIVERSITY OF NEW SOUTH WALES Thesis/Dissertation Sheet Surname or Family name: Platt First name: Edward Other name/s: Richard Murray Abbreviation for degree as grven an the University calendar: MPhil School: Biological, Earth and Environmental Sciences F acuity: Science Title:Mr My thesis addressed two questions: whether survival was inferred to have improved for fish that moved onto land, and what the relative role of predation and density were for detenmining life history variation among populations within one of these land species. For the first question I used life history theory to examine whether survival was inferred to have improved in two fish families which have independently made the transition onto land: Gobiidae and Blenniidae. I examined growth and various aspects of reproductive investment among terrestrial and aquatic species, finding that differences varied according to the level of independence from water. This was consistent with improved survival for certain age classes on land. Nevertheless, the details of life history change differed in each family, with the greatest increases in survival implied for early age classes in Blenniidae, but older age classes in Gobiidae. This suggests fundamental differences in the way the colonization of land occurred in each family. For the second question I investigated the consequences of predataon and density on life hastory variation among frve populations of the Pacific leaping blenny A/ficus amoldorum. -

BMC Evolutionary Biology
Convergent evolution, habitat shifts and variable diversification rates in the ovenbird- woodcreeper family (Furnariidae) Irestedt, M.; Fjeldså, Jon; Dalén, L.; Ericson, P.G.P. Published in: BMC Evolutionary Biology DOI: 10.1186/1471-2148-9-268 Publication date: 2009 Document version Publisher's PDF, also known as Version of record Citation for published version (APA): Irestedt, M., Fjeldså, J., Dalén, L., & Ericson, P. G. P. (2009). Convergent evolution, habitat shifts and variable diversification rates in the ovenbird-woodcreeper family (Furnariidae). BMC Evolutionary Biology, 9(268). https://doi.org/10.1186/1471-2148-9-268 Download date: 03. okt.. 2021 BMC Evolutionary Biology BioMed Central Research article Open Access Convergent evolution, habitat shifts and variable diversification rates in the ovenbird-woodcreeper family (Furnariidae) Martin Irestedt*1, Jon Fjeldså2, Love Dalén1 and Per GP Ericson3 Address: 1Molecular Systematics Laboratory, Swedish Museum of Natural History, PO Box 50007, SE-10405 Stockholm, Sweden, 2Zoological Museum, University of Copenhagen, Universitetsparken 15, DK-2100 Copenhagen, Denmark and 3Department of Vertebrate Zoology, Swedish Museum of Natural History, PO Box 50007, SE-10405 Stockholm, Sweden Email: Martin Irestedt* - [email protected]; Jon Fjeldså - [email protected]; Love Dalén - [email protected]; Per GP Ericson - [email protected] * Corresponding author Published: 21 November 2009 Received: 22 January 2009 Accepted: 21 November 2009 BMC Evolutionary Biology 2009, 9:268 doi:10.1186/1471-2148-9-268 This article is available from: http://www.biomedcentral.com/1471-2148/9/268 © 2009 Irestedt et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. -

A New Genus and Species of Furnariid (Aves: Furnariidae) from the Cocoa-Growing Region of Southeastern Bahia, Brazil
THEWILSONBULLETIN A QUARTERLY MAGAZINE OF ORNITHOLOGY Published by the Wilson Ornithological Society VOL. 108, No. 3 SEPTEMBER 1996 PAGES 397-606 Wilson Bull., 108(3), 1996, pp. 397-433 A NEW GENUS AND SPECIES OF FURNARIID (AVES: FURNARIIDAE) FROM THE COCOA-GROWING REGION OF SOUTHEASTERN BAHIA, BRAZIL Jose FERNANDO PACHECO,’ BRET M. WHITNEY,‘J AND LUIZ P. GONZAGA’ ABSTRACT.-we here describe Acrobatomis fonsecai, a new genus and species in the Furnariidae, from the Atlantic Forest of southeastern Bahia, Brazil. Among the outstanding features of this small, arboreal form are: black-and-gray definitive plumage lacking any rufous: juvenal plumage markedly different from adult; stout, bright-pink legs and feet; and its acrobatic foraging behavior involving almost constant inverted hangs on foliage and scansorial creeping along the undersides of canopy limbs. Analysis of morphology, vocal- izations, and behavior suggest to us a phylogenetic position close to Asfhenes and Crani- oleuca; in some respects, it appears close to the equally obscure Xenerpesres and Meto- pothrix. New data on the morphology, vocalizations, and behavior of several furuariids possibly related to Acrobatornis are presented in the context of intrafamilial relationships. We theorize that Acrobatornis could have colonized its current range during an ancient period of continental semiaridity that promoted the expansion of stick-nesting prototypes from a southern, Chaco-PatagonianE’antanal center, and today represents a relict that sur- vived by adapting to build its stick-nest in the relatively dry, open, canopy of leguminaceous trees of the contemporary humid forest in southeastern Bahia. Another theory of origin places emphasis on the fact that the closest relatives of practically all (if not all) other birds syntopic with Acrobatomis are of primarily Amazonian distribution. -

Integrative and Comparative Biology Advance Access Published May 23, 2013 Integrative and Comparative Biology Integrative and Comparative Biology, Pp
Integrative and Comparative Biology Advance Access published May 23, 2013 Integrative and Comparative Biology Integrative and Comparative Biology, pp. 1–12 doi:10.1093/icb/ict052 Society for Integrative and Comparative Biology SYMPOSIUM Thrash, Flip, or Jump: The Behavioral and Functional Continuum of Terrestrial Locomotion in Teleost Fishes Alice C. Gibb,1,* Miriam A. Ashley-Ross† and S. Tonia Hsieh‡ *Department of Biology, Northern Arizona University, Flagstaff, AZ, USA; †Department of Biology, Wake Forest University, Winston-Salem, NC, USA; ‡Department of Biology, Temple University, Philadelphia, PA, USA From the symposium ‘‘Vertebrate Land Invasions – Past, Present, and Future’’ presented at the annual meeting of the Society for Integrative and Comparative Biology, January 3–7, 2013 at San Francisco, California. Downloaded from 1E-mail: [email protected] Synopsis Moving on land versus in water imposes dramatically different requirements on the musculoskeletal system. http://icb.oxfordjournals.org/ Although many limbed vertebrates, such as salamanders and prehistoric tetrapodomorphs, have an axial system special- ized for aquatic locomotion and an appendicular system adapted for terrestrial locomotion, diverse extant teleosts use the axial musculoskeletal system (body plus caudal fin) to move in these two physically disparate environments. In fact, teleost fishes living at the water’s edge demonstrate diversity in natural history that is reflected in a variety of terrestrial behaviors: (1) species that have only incidental contact -

SICB 2020 Annual Meeting Abstracts
The Society for Integrative and Comparative Biology with the American Microscopical Society The Crustacean Society SICB 2020 ABSTRACT BOOK January 3-7, 2020 JW Marriott Austin • Austin, TX Abstract Book SICB does not assume responsibility for any inconsistencies or errors in the abstracts for contributed paper and poster presentations. We regret any possible omissions, changes and/ or additions not reflected in this abstract book. SICB 2020 Annual Meeting Abstracts 127-1 AARON GOODMAN, AMG*; LAUREN ESPOSITO, LAE; 56-5 ABBOTT, EA*; DIXON, GB; MATZ, MV; University of California Academy of Sciences, California Academy of Sciences ; Texas; [email protected] [email protected] Disentangling coral stress and bleaching responses by comparing Spatial and Ecological Niche Partitioning in Congeneric Scorpions gene expression in symbiotic partners Species in the scorpion genus Centruroides Marx, 1890 (Scorpiones: Coral bleaching—the disruption of the symbiosis between a coral Buthidae) are good candidates to study ecological niche partitioning host and its endosymbiotic algae—is associated with environmental due to their habitat plasticity, widespread geographic distribution, stressors. However, the molecular processes are not well understood, and presence of cryptic species. Currently, three species belonging to and no studies have disentangled the transcriptional bleaching three subgroups of Centruroides are distributed along the Isthmus of response from the stress response. In order to characterize general Tehuantepec in southern Mexico, presenting a rare opportunity to stress response, specific stress responses, and the bleaching response, study niche partitioning within a single genus. We examined the we isolated host and symbiont RNA from fragments of Acropora environmental, substrate, and habitat preferences of Centruroides millepora which were exposed to 5 different stress treatments.