The Expanding Role of Mass Spectrometry in Biotechnology Second Edition
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Recent Progress in Laser Analytics
KOLUMNE 417 CHIMIA 44 (1990) Nr.I~ (Ikzem""r) Chimia 44 (/990) 417 424 <&') Schll'ei=. Chemiker- Verhand; ISSN 0009 4293 Recent Progress in Laser Analytics Analytical methods are on their way to mass spectrometry (MS). In 1946, William penetrate the biological sciences. In this E. Stephens (University of Pennsylvania in trend, laser technology plays an important Philadelphia) described a mass spectrome- role, especially in the form of laser-desorp- ter with time dispersion, followed by the tion mass spectrometry (LD-MS). ion velocitron of A.E. Cameron and D.£. The remarkable progress made in this Eggers. These devices represented early field is nicely demonstrated by a statement forms of the time-of-flight mass spectrom- made in 1986 by Frank H. Field, a specialist eter (TOF-MS) first described by the Swiss in the mass spectrometric investigation of R. Keller in 1949. biomolecules and Professor at Rockefeller The first commercially successful TOF- University in New York. Citing Professor MS was introduced by Bendix Corpora- In dieser Kalwnne schreibl Field: 'The mass region of real interest for tion, and it was based on the design re- Prof Dr. H. M. Widmer proteins lies between 40000 and 100000 ported in 1955 by William C. Wile)' and Forse/lUng Analylik Da, and one can only speculate as to l. H. McLaren (Bendix Aviation Corpom- Ciha-Geigy AG. FO 3.2 CH 4{)()2 Basel whether such monster gaseos ions could be tion). In these early days of TOF-MS, the regelmiissig eigene Meinungsarlike/ oder liidl Giiste produced. My personal feeling is that to do ions were generated by electron impact ein. -

20.5 Ion Detectors
20.5 Ion Detectors • the Faraday cup neutralizes ions to provide a high-precision measurement • the electron multiplier generates a pulse of current for each ion • the photon multiplier uses conversion dynodes to monitor both positive and negative ions • the two multipliers are capable of fast measurements, thus compatible with TOF or Fourier transform methods 20.5 : 1/4 Faraday Cup slit Ions exiting the analyzer are slowed by Faraday cup a positive potential and impact the ion + grounded electrode. At the electrode beam + they are __________ by electrons that RL travel through the load resistor. This ion suppressor causes a voltage across the load resistor that is measured by a high input impedance voltmeter. The impacted electrode is at an angle so that any secondary ions that are created are _____________ by the grounded wall. Of all ion detectors, the Faraday cup provides the most precise relationship between the number of ions exiting the analyzer and the electrical measurement. It is used in __________ experiments such as isotope dilution. Because very small currents are measured, the Faraday cup cannot be used for experiments involving rapid scanning. 20.5 : 2/4 Electron Multiplier ion beam An electron multiplier is constructed just like a photomultiplier but without 16e- 4e- 3 - a _______________. It has dynodes 10 e RL constructed from a Cu/Be alloy, which dynodes is a good secondary electron emitter. +V The dynode potentials are such that the first is grounded and each subsequent one is 100 V more positive. This makes the voltage measurement a little tricky because one end of the load resistor is connected to positive _____________. -

Contents • Introduction to Proteomics and Mass Spectrometry
Contents • Introduction to Proteomics and Mass spectrometry - What we need to know in our quest to explain life - Fundamental things you need to know about mass spectrometry • Interfaces and ion sources - Electrospray ionization (ESI) - conventional and nanospray - Heated nebulizer atmospheric pressure chemical ionization - Matrix assisted laser desorption • Types of MS analyzers - Magnetic sector - Quadrupole - Time-of-flight - Hybrid - Ion trap In the next step in our quest to explain what is life The human genome project has largely been completed and many other genomes are surrendering to the gene sequencers. However, all this knowledge does not give us the information that is needed to explain how living cells work. To do that, we need to study proteins. In 2002, mass spectrometry has developed to the point where it has the capacity to obtain the "exact" molecular weight of many macromolecules. At the present time, this includes proteins up to 150,000 Da. Proteins of higher molecular weights (up to 500,000 Da) can also be studied by mass spectrometry, but with less accuracy. The paradigm for sequencing of peptides and identification of proteins has changed – because of the availability of the human genome database, peptides can be identified merely by their masses or by partial sequence information, often in minutes, not hours. This new capacity is shifting the emphasis of biomedical research back to the functional aspects of cell biochemistry, the expression of particular sets of genes and their gene products, the proteins of the cell. These are the new goals of the biological scientist: o to know which proteins are expressed in each cell, preferably one cell at a time o to know how these proteins are modified, information that cannot necessarily be deduced from the nucleotide sequence of individual genes. -

Modern Mass Spectrometry
Modern Mass Spectrometry MacMillan Group Meeting 2005 Sandra Lee Key References: E. Uggerud, S. Petrie, D. K. Bohme, F. Turecek, D. Schröder, H. Schwarz, D. Plattner, T. Wyttenbach, M. T. Bowers, P. B. Armentrout, S. A. Truger, T. Junker, G. Suizdak, Mark Brönstrup. Topics in Current Chemistry: Modern Mass Spectroscopy, pp. 1-302, 225. Springer-Verlag, Berlin, 2003. Current Topics in Organic Chemistry 2003, 15, 1503-1624 1 The Basics of Mass Spectroscopy ! Purpose Mass spectrometers use the difference in mass-to-charge ratio (m/z) of ionized atoms or molecules to separate them. Therefore, mass spectroscopy allows quantitation of atoms or molecules and provides structural information by the identification of distinctive fragmentation patterns. The general operation of a mass spectrometer is: "1. " create gas-phase ions "2. " separate the ions in space or time based on their mass-to-charge ratio "3. " measure the quantity of ions of each mass-to-charge ratio Ionization sources ! Instrumentation Chemical Ionisation (CI) Atmospheric Pressure CI!(APCI) Electron Impact!(EI) Electrospray Ionization!(ESI) SORTING DETECTION IONIZATION OF IONS OF IONS Fast Atom Bombardment (FAB) Field Desorption/Field Ionisation (FD/FI) Matrix Assisted Laser Desorption gaseous mass ion Ionisation!(MALDI) ion source analyzer transducer Thermospray Ionisation (TI) Analyzers quadrupoles vacuum signal Time-of-Flight (TOF) pump processor magnetic sectors 10-5– 10-8 torr Fourier transform and quadrupole ion traps inlet Detectors mass electron multiplier spectrum Faraday cup Ionization Sources: Classical Methods ! Electron Impact Ionization A beam of electrons passes through a gas-phase sample and collides with neutral analyte molcules (M) to produce a positively charged ion or a fragment ion. -
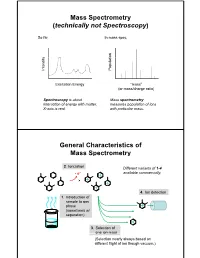
Mass Spectrometry (Technically Not Spectroscopy)
Mass Spectrometry (technically not Spectroscopy) So far, In mass spec, on y Populati Intensit Excitation Energy “mass” (or mass/charge ratio) Spectroscopy is about Mass spectrometry interaction of energy with matter. measures population of ions X-axis is real. with particular mass. General Characteristics of Mass Spectrometry 2. Ionization Different variants of 1-4 -e- available commercially. 4. Ion detection 1. Introduction of sample to gas phase (sometimes w/ separation) 3. Selection of one ion mass (Selection nearly always based on different flight of ion though vacuum.) General Components of a Mass Spectrometer Lots of choices, which can be mixed and matched. direct injection The Mass Spectrum fragment “daughter” ions M+ “parent” mass Sample Introduction: Direc t Inser tion Prob e If sample is a liquid, sample can also be injected directly into ionization region. If sample isn’t pure, get multiple parents (that can’t be distinguished from fragments). Capillary Column Introduction Continous source of molecules to spectrometer. detector column (including GC, LC, chiral, size exclusion) • Signal intensity depends on both amount of molecule and ionization efficiency • To use quantitatively, must calibrate peaks with respect eltilution time ttlitotal ion curren t to quantity eluted (TIC) over time Capillary Column Introduction Easy to interface with gas or liquid chromatography. TIC trace elution time time averaged time averaged mass spectrum mass spectrum Methods of Ionization: Electron Ionization (EI) 1 - + - 1 M + e (kV energy) M + 2e Fragmentation in Electron Ionization daughter ion (observed in spectrum) neutral fragment (not observed) excited parent at electron at electron energy of energy of 15 eV 70 e V Lower electron energy yields less fragmentation, but also less signal. -

An Introduction to Mass Spectrometry
An Introduction to Mass Spectrometry by Scott E. Van Bramer Widener University Department of Chemistry One University Place Chester, PA 19013 [email protected] http://science.widener.edu/~svanbram revised: September 2, 1998 © Copyright 1997 TABLE OF CONTENTS INTRODUCTION ........................................................... 4 SAMPLE INTRODUCTION ....................................................5 Direct Vapor Inlet .......................................................5 Gas Chromatography.....................................................5 Liquid Chromatography...................................................6 Direct Insertion Probe ....................................................6 Direct Ionization of Sample ................................................6 IONIZATION TECHNIQUES...................................................6 Electron Ionization .......................................................7 Chemical Ionization ..................................................... 9 Fast Atom Bombardment and Secondary Ion Mass Spectrometry .................10 Atmospheric Pressure Ionization and Electrospray Ionization ....................11 Matrix Assisted Laser Desorption/Ionization ................................ 13 Other Ionization Methods ................................................13 Self-Test #1 ...........................................................14 MASS ANALYZERS .........................................................14 Quadrupole ............................................................15 -

Methods of Ion Generation
Chem. Rev. 2001, 101, 361−375 361 Methods of Ion Generation Marvin L. Vestal PE Biosystems, Framingham, Massachusetts 01701 Received May 24, 2000 Contents I. Introduction 361 II. Atomic Ions 362 A. Thermal Ionization 362 B. Spark Source 362 C. Plasma Sources 362 D. Glow Discharge 362 E. Inductively Coupled Plasma (ICP) 363 III. Molecular Ions from Volatile Samples. 364 A. Electron Ionization (EI) 364 B. Chemical Ionization (CI) 365 C. Photoionization (PI) 367 D. Field Ionization (FI) 367 IV. Molecular Ions from Nonvolatile Samples 367 Marvin L. Vestal received his B.S. and M.S. degrees, 1958 and 1960, A. Spray Techniques 367 respectively, in Engineering Sciences from Purdue Univesity, Layfayette, IN. In 1975 he received his Ph.D. degree in Chemical Physics from the B. Electrospray 367 University of Utah, Salt Lake City. From 1958 to 1960 he was a Scientist C. Desorption from Surfaces 369 at Johnston Laboratories, Inc., in Layfayette, IN. From 1960 to 1967 he D. High-Energy Particle Impact 369 became Senior Scientist at Johnston Laboratories, Inc., in Baltimore, MD. E. Low-Energy Particle Impact 370 From 1960 to 1962 he was a Graduate Student in the Department of Physics at John Hopkins University. From 1967 to 1970 he was Vice F. Low-Energy Impact with Liquid Surfaces 371 President at Scientific Research Instruments, Corp. in Baltimore, MD. From G. Flow FAB 371 1970 to 1975 he was a Graduate Student and Research Instructor at the H. Laser Ionization−MALDI 371 University of Utah, Salt Lake City. From 1976 to 1981 he became I. -

Hydrogen Abstraction from Titanium and Zirconium Oxides
The Pennsylvania State University The Graduate School Eberly College of Science NEW INSIGHTS IN THE GAS-PHASE ION CHEMISTRY OF GROUP IVB TRANSITION METAL OXIDES: CLUSTER MODELS FOR CATALYSIS A Dissertation in Chemistry by Christopher L. Harmon © 2016 Christopher L. Harmon Submitted in Partial Fulfillment Of the Requirements For the Degree of Doctor of Philosophy August 2016 The dissertation of Christopher L. Harmon was reviewed and approved* by the following: A. W. Castleman, Jr. Evan Pugh Professor of Chemistry and Physics Eberly Distinguished Chair in Science Dissertation Adviser Chair of Committee Nicholas Winograd Evan Pugh Professor of Chemistry Lasse Jensen Associate Professor of Chemistry Robert M. Rioux Friedrich G. Helfferich Associate Professor of Chemical Engineering Professor of Chemistry Kenneth S. Feldman Professor of Chemistry Graduate Program Chair *Signatures are on file in the Graduate School. ii Abstract The study of gas-phase cluster models has emerged as a useful tool to model catalytic active sites on surfaces. By observing how the reactivity of TMO clusters changes with cluster size, active site, and metal composition, we draw insights about the molecular factors governing reactivity and selectivity. The aim is to provide a conceptual framework to drive the development of improved catalysts with higher activity and selectivity. As group IVB transition metal oxides (TMOs) are widely used as catalysts and supports, we have undertaken mass spectrometric reactivity studies of titanium, zirconium, and hafnium oxide clusters with small organic molecules of interest in catalysis. We particularly note the differences in reactivity observed as the identity of the + metal in the cluster changes. We report the reactivity of MxO2x (M = Ti, Zr) clusters that contain radical atomic oxygen, O•−, with methane, ethane, propane, ethylene, and propylene. -

Analytical Chemistry, Strategic Exercise: Organic Aerosol Analysis
WIR SCHAFFEN WISSEN –HEUTE FÜR MORGEN Analytical Chemistry, Strategic exercise: Organic aerosol analysis Urs Baltensperger Laboratory of Atmospheric Chemistry, Paul Scherrer Institute, Villigen , Switzerland ETHZ, HS 2020, 8.12.2020 Aerosol size distribution, and sources and sinks Chemical composition of PM1 in Zurich in winter Energiespiegel 19/2008 http://gabe.web.psi.ch/pdfs/Energiespiegel_19_e.pdf Today’s task: Organic aerosol analysis Page 4 The state of the art in 2009 Hallquist et al., ACP 2009 Traditional methods can identify only a fraction of organics Rogge et al., 1993 Most detailed analysis performed with off-line methods: More than 10‘000 different organic compounds isolated with GCxGC-TOF/MS Hamilton et al., 2004 NB: isolated ≠ identified…. Besides extraction, also thermal desorption is applied: TAG (Goldstein et al., 2008) Organic acids formed in the photooxidation of -pinene All acids found in aerosol phase. Some of them are highly volatile (e.g., formic, acetic acid) and not expected to be there. Probably hydrolysis of oligomers Fisseha et al., Anal. Chem. 2004 Developments since 2009 EESI CHARON‐ PTRMS FIGAERO -I-CIMS Adapted from Hallquist et al., 2009 9 Required for mass spectrometry: ionization of individual molecules • For gas phase: relatively easy (avoid fragmentation, clustering) • For aerosol particles: Much more difficult: either dissolution in a solvent or evaporation - Problem with dissolution: only the soluble fraction accessible - Problem with evaporation: fragmentation Page 10 What is Mass Spectrometry -

QMG 422 Analyzers
Operating Instructions Incl. Declaration of Conformity QMG 422 Analyzers BG 805 983 BE (0112) 1 Product Identification In all communications with Pfeiffer Vacuum, please specify the information on the product nameplate. For convenient reference copy that information into the space provided below. Pfeiffer Vacuum, D-35614 Asslar Typ: No: F-No: Validity This document applies to the QMA 400, QMA 410, QMA 430 with Faraday cup or 90° off-axis SEM and Faraday cup and with the ion sources described in this document. Illustrations If not indicated otherwise in the legends, the illustrations in this document corre- spond to the QMA 400 with 90° off-axis SEM. They apply to other types by analogy. Designations The short designation "QMA" is used for the QMA 400, QMA 410 and QMA 430. The short designation "QMH" is used for the QMH 400 and QMH 410. In contrast to the operating instructions for the QMG 422, these instructions follow the same conventions as the Pfeiffer QuadStar™ 422 operating instructions. The parameters are marked with quotation marks ("…") e.g. "Resolution". Technical changes We reserve the right to make technical changes without prior notice. Intended Use The QMA 400, QMA 410 and QMA 430 Analyzers are used for gas analysis in high vacuum. They are part of the QMG 422 mass spectrometer system and may only be used in connection with equipment belonging to that system. The operating instructions of all system components must be strictly followed. 2 BG 805 983 BE (0112) QMA4x0.oi Contents Product Identification 2 Validity 2 Intended -

Selective Chemical Ionization of Nitrogen and Sulfur Heterocycles in Petroleum Fractions by Ion Trap Mass Spectrometry
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Selective Chemical Ionization of Nitrogen and Sulfur Heterocycles in Petroleum Fractions by Ion Trap Mass Spectrometry C. S. Creaser*, F. Krokos, and K. E. O’Neill Shod of Chemical Sciences, University of East Anglia, Norwich, United Kingdom M. J. C. Smith and P. G. McDowell BP Research, Sunbury-on-Thames, Middlesex, United Kingdom A procedure is reported for the selective ammonia chemical ionization of some nitrogen and sulfur heterocycles in petroleum fractions using ion trap mass spectrometry (ITMS). The ion trap scan routine is designed to optimize the population of ammonium reagent ions and eject from the trap (by radio frequency/direct current isolation) electron ionization products formed during reagent ion formation prior to ionization of the sample. The ITMS procedure is compared with standard ion trap detector and conventional quadrupole ammonia chemi- cal ionization for the determination of nitrogen and sulfur heterocycles in gas oil and kerosine samples. Greatly enhanced selectivity is shown for the ITMS procedure by sup res- sion of competing charge-exchange processes. (1 Am Sot Mass Spectrum 1993, 4, 322-326 ‘; he removal of organic compounds containing reagent ion NH;, is formed via the reactions [5] nitrogen and sulfur is an important part of the T petroleum-refining process 111. Their presence in NH, ‘1 NH;’ (1) petroleum distillates results in the formation of envi- NH, f NH:‘+ NH; + NH; (2) ronmental pollutants (SO,, NO,) during combustion, and their reactivity can lead to catalyst poisoning Whereas NH: is unreactive toward a wide range of in refinery processes. -

Electron Microscopy Are the Beam Damaging Effects That Can Arise from Interactions Between the Primary High Energy Electron Beam and the Sample
Thermal in situ TEM experiment on nanoscale indium islands Estimation of Electron Beam Heating Effect Master’s thesis in Applied Physics REN QIU Department of Physics CHALMERS UNIVERSITY OF TECHNOLOGY Gothenburg, Sweden 2017 Master’s thesis 2017 Thermal in situ TEM experiment on nanoscale indium islands Estimation of Electron Beam Heating Effect REN QIU Department of Physics Eva Olsson Group Chalmers University of Technology Gothenburg, Sweden 2017 Thermal in situ TEM Experiment on Nanoscale Indium Islands Estimation of Electron Beam Heating Effect Master Thesis within the Masters Programme of Applied Physics © REN QIU, 2017. Supervisor: Eva Olsson, Norvik Voskanian, Lunjie Zeng, Department of Physics Examiner: Eva Olsson, Department of Physics Master’s Thesis 2017 Department of Physics Eva Olsson Group Chalmers University of Technology SE-412 96 Gothenburg Telephone + 46(0)73-732 4883 Cover: Top left: Indium islands segmentation by watershed algorithm based on the bright field TEM image. Top right: Dark field image of the in situ melting experiment where the indium islands with bright pixels are melted and the ones with dark pixels are in solid state. Bottom left: Nano-scale gold tip fabricated through electrochemical etching. Bottom right: A simple Faraday cup made on the gold tip by focus ion beam (FIB) method. Printed by Chalmers Reproservice Gothenburg, Sweden 2017 iv Thermal in situ TEM Experiment on Nanoscale Indium Islands Estimation of Electron Beam Heating Effect REN QIU Department of Physics Chalmers University of Technology Abstract One of the limitations of transmission electron microscopy are the beam damaging effects that can arise from interactions between the primary high energy electron beam and the sample.