Reversibility of Ferri-/Ferrocyanide Redox During Operando Soft X-Ray Spectroscopy
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Excess Heat Production in the Redox Couple Reaction of Ferricyanide and Ferrocyanide
www.nature.com/scientificreports OPEN Excess heat production in the redox couple reaction of ferricyanide and ferrocyanide Atsushi Sugiyama1,2,3*, Makoto Miura4, Yoshinobu Oshikiri5, Yena Kim3, Ryoichi Morimoto6, Miki Miura7, Tetsuya Osaka2, Iwao Mogi8, Yusuke Yamauchi3,9* & Ryoichi Aogaki3,10* In order to establish the universality of the excess heat production in electrochemical reaction, under a high magnetic feld, as one of the most fundamental electrochemical reactions, the case of ferricyanide-ferrocyanide redox reaction was examined, where ionic vacancies with ± 1 unit charge were collided by means of magnetohydrodynamic (MHD) fow. As a result, from the pair annihilation of the vacancies with opposite signs, beyond 7 T, excess heat production up to 25 kJ·mol−1 in average at 15 T was observed, which was attributed to the liberation of the solvation energy stored in a pair of the vacancy cores with a 0.32 nm radius, i.e., 112 kJ·mol−1. Diference between the observed and expected energies comes from the small collision efciency of 0.22 due to small radius of the vacancy core. Ionic vacancy initially created as a by-product of electrode reaction is unstable in solution phase, stabilized by releasing solvation energy. Ionic vacancy utilizes the energy to enlarge the core and stores the energy in it. As a result, solvated ionic vacancy consists of a polarized free space of the enlarged core surrounded by oppositely charged ionic cloud. The accuracy and precision of the measured values were ascertained by in situ standard additive method. Recently, in copper redox reaction under a high magnetic feld, great excess heat production up to 410 kJ·mol−1 in average has been observed, which is 1.5 times larger than the molar combustion heat of hydrogen (285.8 kJ·mol−1)1. -

United States Patent 19 11 Patent Number: 5,360,712 Olm Et Al
D US005360712A United States Patent 19 11 Patent Number: 5,360,712 Olm et al. 45 Date of Patent: Nov. 1, 1994 54 INTERNALLY DOPED SILVER HALIDE 4,981,781 1/1991 McDugle et al. ................... 430/605 EMULSIONS AND PROCESSES FOR THER 5,037,732 8/1991 McDugle et al. ................... 430/567 ARA 5,132,203 7/1992 Bell et al. ............. ... 430/567 PREP TION 5,268,264 12/1993 Marchetti et al. .................. 430/605 75) Inventors: McDugle;Myra T. Olm, Sherrin Webster; A. Puckett, Woodrow both G.of FOREIGN PATENT DOCUMENTS Rochester; Traci Y. Kuromoto, West 513748A1 11/1992 European Pat. Off. ... GO3C 7/392 Henrietta; Raymond S. Eachus, Rochester; Eric L. Bell, Wesbter; OTHER PUBLICATIONS Robert D. Wilson, Rochester, all of Research Disclosure, vol. 176, Dec. 1978, Item 17643, N.Y. Section I, subsection A. Research Disclosure, vol. 308, Dec. 1989, Item 308119, 73) Assignee: Eastman Kodak Company, Section, I, subsection D. Rochester, N.Y. 21 Appl. No.: 91,148 Primary Examiner-Janet C. Baxter J. v. V. 19 Attorney, Agent, or Firm-Carl O. Thomas 51) Int. Cl................................................. GC/ A process is disclosed of preparing a radiation sensitive 52 U.S.C. .................................... 430/567; 430/569; silver halide emulsion comprising reacting silver and 430/604; 430/605 halide ions in a dispersing medium in the presence of a 58 Field of Search ................ 430/567, 569, 604, 605 metal hexacoordination or tetracoordination complex (56) References Cited having at least one organic ligand containing a least one carbon-to-carbon bond, at least one carbon-to-hydro U.S. -
![Determination of the Activation Parameters of Reaction Between [Fe(CN6] and K[Co(HEDTA)NO2]." (2009)](https://docslib.b-cdn.net/cover/8644/determination-of-the-activation-parameters-of-reaction-between-fe-cn6-and-k-co-hedta-no2-2009-998644.webp)
Determination of the Activation Parameters of Reaction Between [Fe(CN6] and K[Co(HEDTA)NO2]." (2009)
East Tennessee State University Digital Commons @ East Tennessee State University Electronic Theses and Dissertations Student Works 12-2009 Determination of the Activation Parameters of -4 Reaction Between [Fe(CN6] and K[Co(HEDTA)NO2]. Sammy Eni Eni East Tennessee State University Follow this and additional works at: https://dc.etsu.edu/etd Part of the Organic Chemistry Commons Recommended Citation -4 Eni Eni, Sammy, "Determination of the Activation Parameters of Reaction Between [Fe(CN6] and K[Co(HEDTA)NO2]." (2009). Electronic Theses and Dissertations. Paper 1798. https://dc.etsu.edu/etd/1798 This Thesis - Open Access is brought to you for free and open access by the Student Works at Digital Commons @ East Tennessee State University. It has been accepted for inclusion in Electronic Theses and Dissertations by an authorized administrator of Digital Commons @ East Tennessee State University. For more information, please contact [email protected]. -4 The Determination of the Activation Parameters of Reaction Between [Fe(CN)6] and K[Co(HEDTA)NO2] ________________________________ A thesis presented to the faculty of the Department of Chemistry East Tennessee State University in Partial Fulfillment of the requirements for the degree Master of Science in Chemistry ________________________________ by Sammy Eni Eni December 2009 ________________________________ Dr. Jeffrey Wardeska, Chair Dr. Ningfeng Zhao Dr. Yu-Lin Jiang -4 Keywords: Activation parameters, [Fe(CN)6] , K[Co(HEDTA)NO2]. ABSTRACT -4 Determination of the Activation Parameters of Reaction Between [Fe(CN)6] and K[Co(HEDTA)NO2] by Sammy Eni Eni -4 The kinetics of the oxidation of [Fe(CN)6] by K[Co(HEDTA)NO2] was studied in order to get the mechanism and the activation parameters of the reaction. -

Investigation of Novel Nanoparticles of Gallium Ferricyanide and Gallium Lawsonate As Potential Anticancer Agents, and Nanoparti
Investigation of Novel Nanoparticles of Gallium Ferricyanide and Gallium Lawsonate as Potential Anticancer Agents, and Nanoparticles of Novel Bismuth Tetrathiotungstate as Promising CT Contrast Agent A Thesis submitted to Kent State University In partial fulfillment of the requirements for the degree of Master of Science Liu Yang August 2014 Thesis written by Liu Yang B.S. Kent State University, 2013 M.S. Kent State University, 2014 Approved by ___________________________________, Advisor, Committee member Dr. Songping Huang ___________________________________, Committee member Dr. Scott Bunge ___________________________________, Committee member Dr. Mietek Jaroniec Accepted by ___________________________________, Chair, Department of Chemistry Dr. Michael Tubergen ___________________________________, Dean, College of Arts and Sciences Dr. James L. Blank ii Table of Contents List of Figures..…………………………………………………………………........vii Acknowledgements ……………………………………………………………….….xi Chapter 1: Summary, Materials and Methods …..……………………………………1 1.1 Materials ………………………………………………………………….3 1.1.1 carboxymethyl reduced polysaccharide (CMRD) preparation….3 1.2 Methods …………………………………………………………………4 1.2.1 Atomic absorption spectroscopy (AA) …………………………4 1.2.2 Acid base treating method ……………………………………...4 1.2.3 Cell viability study ……………………………………………...5 i) MTT assay…………………………………………………..5 ii) Trypan blue assay ………………………………………….6 1.2.4 Dialysis …………………………………………………………6 1.2.5 Elementary analysis …………………………………………….7 1.2.6 Lyophilization …………………………………………………..7 iii 1.2.7 -
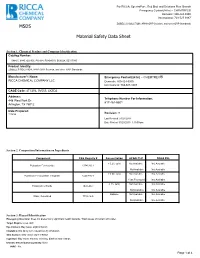
MSDS Material Safety Data Sheet
For RICCA, SpectroPure, Red Bird, and Solutions Plus Brands Emergency Contact(24 hr) -- CHEMTREC® Domestic: 800-424-9300 International: 703-527-3887 ZOBELL'S SOLUTION, APHA ORP Solution, and other ORP Standards MSDS Material Safety Data Sheet Section 1: Chemical Product and Company Identification Catalog Number: 5464.5, 9880, OX-905, PX-918, R5464510, S0932A, SZ117300 Product Identity: ZOBELL'S SOLUTION, APHA ORP Solution, and other ORP Standards Manufacturer's Name: Emergency Contact(24 hr) -- CHEMTREC® RICCA CHEMICAL COMPANY LLC Domestic: 800-424-9300 International: 703-527-3887 CAGE Code: 4TCW6, 0V553, 4XZQ2 Address: Telephone Number For Information: 448 West Fork Dr 817-461-5601 Arlington, TX 76012 Date Prepared: 11/9/98 Revision: 9 Last Revised: 01/24/2014 Date Printed: 03/25/2015 1:19:09 pm Section 2. Composition/Information on Ingredients Component CAS Registry # Concentration ACGIH TLV OSHA PEL < 0.2% (w/v) Not Available Not Available Potassium Ferricyanide 13746-66-2 Not Available Not Available < 0.5% (w/v) Not Available Not Available Potassium Ferrocyanide Trihydrate 14459-95-1 1 (as Fe) mg/m3 Not Available < 1% (w/v) Not Available Not Available Potassium Chloride 7447-40-7 Not Available Not Available Balance Not Available Not Available Water, Deionized 7732-18-5 Not Available Not Available Section 3: Hazard Identification Emergency Overview: Does not present any significant health hazards. Wash areas of contact with water. Target Organs: eyes, skin Eye Contact: May cause slight irritation. Inhalation: Not likely to be hazardous by inhalation. Skin Contact: May cause slight irritation. Ingestion: May cause nausea, vomiting, diarrhea and cramps. -

No, Potassium Ferrocyanide in Some Salt Brands Is Not at Toxic Levels
Fact check: No, potassium ferrocyanide in some salt brands is not at toxic levels Shubashree Desikan JULY 02, 2019 Citing a report from U.S.-based West American Analytical Laboratories, Shiv Shankar Gupta, chairman of Godhum Grains and Food Products, accused some salt manufacturers in India of selling food-grade salt that contained high levels of potassium ferrocyanide. This caused a panic, which was aggravated by widespread sharing of the news item on social media. Indeed, potassium ferrocyanide is being used in salt to give it anti-caking properties. But the answer to whether it is toxic is no! Firstly, while potassium cyanide is a toxic substance and releases the cyanide anion when consumed by a person, potassium ferrocyanide is not. In potassium ferrocyanide, the cyanide anion is strongly bonded to ferrous ion and hence does not get hydrolysed. An image widely shared in social media. Secondly, there is the factor known as the LD50 value. This is expanded as “Lethal Dose 50%”. LD50 is the amount of any chemical that can cause death in 50% of the group of animals that it enters by consumption or absorption through the skin. According to inorganic chemist Dr. Sayam Sengupta of IISER Kolkata, the LD50 value for common salt — sodium chloride — for rats is greater than 3 gram per kilogram body weight of the animal. He points out that for potassium Ferrocyanide, LD 50 value is 3.6 gram per kilogram weight of the animal, when taken orally. As this indicates, the LD50 value of potassium ferrocyanide is almost the same as common salt. -

On the Cyanogen Halides by P
ON THE CYANOGEN HALIDES BY P. KAILASAM (From the Chemistry Department, Madras Christian College) Received July 31, 1941 (Communicated by Sir C. V. Raman, xt., F.R.S., NN.L.) I. Action of Halogens on Cyanogen Halides No observations appear to have been recorded on the action of halogens on cyanogen halides except that of chlorine on cyanogen chloride (Wurtz, 1851; Naumann and Vogt, 1870) and bromine on cyanogen bromide in ether (Poonamarew, 1885) which polymerise them to cyanuric chloride and cyanuric bromide respectively. The object of the present investigation is to study the possible displacement reactions with the cyanogen halides. The action of chlorine gas on cyanogen iodide and the action of bromine on cyanogen chloride were first studied. It was expected that chlorine would displace iodine from cyanogen iodide forming cyanogen chloride, but it was found that cyanogen iodide was not affected by dry chlorine. Dry cyanogen iodide, prepared by warming an intimate mixture of one part of iodine and two parts of AgCN, at about 40°, was sublimed into a narrow tube which could be heated in a water-bath. Dry chlorine was passed through the tube for nearly two hours. No immediate reaction occurred, and even after leaving for a week at room temperature (30°) the white needles of cyanogen iodide remained unchanged. The tube was then slowly heated, but no reaction occurred up to the sublimation temperature of cyanogen iodide (about 40°). It was found that cyanogen iodide could be sublimed unchanged in an atmosphere of dry chlorine. Since chlorine does not displace iodine from cyanogen iodide, it was thought that bromine might displace chlorine from cyanogen chloride form- ing cyanogen bromide. -

The Light Triggered Dissolution of Gold Wires Using Potassium Ferrocyanide T Solutions Enables Cumulative Illumination Sensing ⁎ Weida D
Sensors & Actuators: B. Chemical 282 (2019) 52–59 Contents lists available at ScienceDirect Sensors and Actuators B: Chemical journal homepage: www.elsevier.com/locate/snb The light triggered dissolution of gold wires using potassium ferrocyanide T solutions enables cumulative illumination sensing ⁎ Weida D. Chena, Seung-Kyun Kangb, Wendelin J. Starka, John A. Rogersc,d,e, Robert N. Grassa, a Department of Chemistry and Applied Biosciences, ETH Zurich, Vladimir-Prelog-Weg 1, 8093 Zurich, Switzerland b Department of Bio and Brain Engineering, KAIST, 291 Daehak-ro, Yuseong-gu, Daejeon 334141, Republic of Korea c Departments of Materials Science and Engineering, Biomedical Engineering, Neurological Surgery, Chemistry, Mechanical Engineering, Electrical Engineering and Computer Science, Northwestern University, Evanston, IL 60208, USA d Center for Bio-Integrated Electronics, Northwestern University, Evanston, IL 60208, USA e Simpson Querrey Institute for Nano/Biotechnology, Northwestern University, Evanston, IL 60208, USA ARTICLE INFO ABSTRACT Keywords: Electronic systems with on-demand dissolution or destruction capabilities offer unusual opportunities in hard- Photochemistry ware-oriented security devices, advanced military spying and controlled biological treatment. Here, the dis- Cyanide solution chemistry of gold, generally known as inert metal, in potassium ferricyanide and potassium ferrocya- Sensor nide solutions has been investigated upon light exposure. While a pure aqueous solution of potassium Conductors ferricyanide–K3[Fe(CN)6] does not dissolve gold, an aqueous solution of potassium ferrocyanide–K4[Fe(CN)6] Diffusion limitation irradiated with ambient light is able to completely dissolve a gold electrode within several minutes. Photo Devices activation and dissolution kinetics were assessed at different initial pH values, light irradiation intensities and ferrocyanide concentrations. -

Long-Term Ferrocyanide Application Via Deicing Salts Promotes the Establishment of Actinomycetales Assimilating Ferrocyanide-Derived Carbon in Soil
bs_bs_banner Long-term ferrocyanide application via deicing salts promotes the establishment of Actinomycetales assimilating ferrocyanide-derived carbon in soil Silvia Gschwendtner,1 Tim Mansfeldt,2 soils, belonging mostly to Actinomycetales Susanne Kublik,1 Evangelia Touliari,1 (Kineosporia, Mycobacterium, Micromonosporaceae). Franz Buegger3 and Michael Schloter1,* In the soil without pre-exposition, bacteria belonging 1Research Unit Environmental Genomics, Helmholtz to Acidobacteria (Gp3, Gp4, Gp6), Gemmatimonade- Zentrum Munchen,€ German Research Center for tes (Gemmatimonas)andGammaproteobacteria Environmental Health (GmbH), Ingolstadter€ Landstraße 1, (Thermomonas, Xanthomonadaceae)usedferro- Neuherberg 85764, Germany. cyanide as C source but not the present Actinomyc- 2Department Geowissenschaften, Bodengeographie/ etales. This indicated that (i) various bacteria are able Bodenkunde, Universitat€ zu Koln,€ Albertus-Magnus- to assimilate ferrocyanide-derived C and (ii) long-term Platz, Koln€ 50923, Germany. exposition to ferrocyanide applied with deicing salts 3Institute of Biochemical Plant Pathology, Helmholtz leads to Actinomycetales outcompeting other microor- Zentrum Munchen,€ German Research Center for ganisms for the use of ferrocyanide as C source. Environmental Health (GmbH), Ingolstadter€ Landstraße 1, Neuherberg 85764, Germany. Introduction Cyanide is produced by various organisms including Summary bacteria, algae, fungi and higher plants as defence Cyanides are highly toxic and produced by various mechanism or offensive strategy (Møller and Seigler, microorganisms as defence strategy or to increase 1998; Gallagher and Manoil, 2001; Zagrobelny et al., their competitiveness. As degradation is the most 2008) and consequently occurs naturally at low levels in efficient way of detoxification, some microbes devel- the environment. However, it is highly toxic for living oped the capability to use cyanides as carbon and organisms by complexing metalloproteins, e.g. -

Problems 14, 15, 16, 19, 22, 24, and 25 in Dekock and Gray, Pages 411–414. 14. “All Octahedral Complexes of V3+ Have The
Problems 14, 15, 16, 19, 22, 24, and 25 in DeKock and Gray, pages 411–414. 14. “All octahedral complexes of V3+ have the same number of unpaired electrons, no matter what the nature of the ligand. Why is this so?” V3+ has a d2 electron configuration; in an octahedral ligand environment, regardless of the type of ligand interactions, the lowest energy orbitals are the triply degenerate 2 t2g orbitals. A d octahedral metal complex will always have 2 unpaired electrons. For the same reason, octahedral d1, d3, d8, d9, and d10 also only have one possible electronic configuration. 15. “How does the molecular-orbital theory account for the order of ligands in the spectrochemical series.” The spectrochemical series (courtesy of Wikipedia): I− < Br− < S2− < SCN− < Cl− < − − − − 2− − NO3 < N3 < F < OH < C2O4 ≈ H2O < NCS < CH3CN < pyridine < NH3 < − − ethylenediamine < 2,2'-bipyridine < 1,10-phenanthroline < NO2 < PPh3 < CN ≈ CO. This series can be explained by the type of π interactions on the ligands. The ligands with the smallest octahedral splitting will be π-donors (i.e. I-, Br-, etc) who will bond with the t2g orbitals on the metal, raising it in energy, thus decreasing the - t2g eg splitting. Similarly, π-acceptors (i.e. CN , CO, etc) will have the largest octahedral splitting energy as the t2g orbitals on the metal now become bonding and decrease in energy. The ligands in the middle of the series have little π-interaction with the metal and therefore the t2g remain nonbonding. 3- 4- 16. “Explain why Co(CN)6 is extremely stable but Co(CN)6 is not.” 3- 6 6 4- 7 6 1 Co(CN)6 is low spin d , therefore t2g , while Co(CN)6 is low spin d , t2g eg* . -

Phenethyl Isothiocyanate (Peitc) Decreases Specificity
PHENETHYL ISOTHIOCYANATE (PEITC) DECREASES SPECIFICITY PROTEIN (SP) TRANSCRIPTION FACTORS THROUGH AN ROS- DEPENDENT MECHANISM A Thesis by AARON SHANE GUTHRIE Submitted to the Office of Graduate Studies of Texas A&M University in partial fulfillment of the requirements for the degree of MASTER OF SCIENCE Approved by: Chair of Committee, Stephen Safe Committee Members, Robert Burghardt Xiuren Zhang Head of Department, Gregory D. Reinhart December 2012 Major Subject: Biochemistry Copyright 2012 Aaron Shane Guthrie ABSTRACT Isothiocyanates (ITCs) are phytochemicals highly expressed in cruciferous vegetables and these compounds are associated with the decreased incidence of cancers in populations consuming high levels of cruciferous vegetables. Several individual ITCs including phenethyl isothiocyanate (PEITC) inhibit tumor growth and angiogenesis and their anticancer activity has been linked to inhibition of cancer cell growth, survival and inflammation (NFB). It has also been demonstrated that PEITC induces reactive oxygen species (ROS) and that ROS is largely responsible for PEITC-induced cell death. To confirm PEITC-induced cancer cell death we have investigated the mechanism of action of PEITC in pancreatic cancer cell lines and PEITC induces ROS and inhibits growth and induces apoptosis (PARP cleavage). In addition, PEITC downregulates expression of several gene products including vascular endothelial growth factor (VEGF), cyclin D1 (CD1), Bcl2 and survivin and these have previously been reported in other studies. However, since these gene products are all regulated by specificity protein (Sp) transcription factors Sp1, Sp3 and Sp4, which are overexpressed in cancer cells and tumors, we investigated the effects of PEITC on Sp proteins and observed that PEITC decreased expression of Sp1, Sp3 and Sp4 in pancreatic cancer cells. -

Recovery of Gold from Ore with Potassium Ferrocyanide Solution Under UV Light
minerals Article Recovery of Gold from Ore with Potassium Ferrocyanide Solution under UV Light Ziyuan Liu 1 , Jue Kou 1,*, Yi Xing 2 and Chunbao Sun 1 1 School of Civil and Resource Engineering, University of Science and Technology, Beijing 100083, China; [email protected] (Z.L.); [email protected] (C.S.) 2 School of Energy and Environmental Engineering, University of Science and Technology, Beijing 100083, China; [email protected] * Correspondence: [email protected] Abstract: In this study, potassium ferrocyanide, a nontoxic cyanide precursor in dark and diffuse reflection environment, was applied as reagent for the leaching of gold. The free cyanide ions could gradually release from potassium ferrocyanide solution under the ultraviolet light. Orthogonal leaching experiments were performed in gold ore to analyze the effect of solution pH, potassium ferrocyanide dosage, and temperature in a potassium ferrocyanide solution system under UV light. Response surface methodology (RSM) was applied to explore the role of potassium ferrocyanide in gold leaching; optimized results showed that the gold recovery reached 67.74% in a high-alkaline envi- ronment at a 12.6 pH, 3.8 kg/t potassium ferrocyanide dosage, 62 ◦C, and irradiance of 10 mW·cm−2. The gold leaching kinetics were monitored by quartz crystal microbalance with dissipation (QCM-D) of potassium ferrocyanide solution. The results indicate that the gold extraction process could be divided into two stages: adsorption and leaching, and a rigid adsorption layer formed on the reaction surface. Furthermore, X-ray photoelectron spectroscopy (XPS) analysis of the gold sensor surface after leaching reaction showed that –C≡N appears on the gold sensor surface, and the gold is Citation: Liu, Z.; Kou, J.; Xing, Y.; oxidized to form AuCN complexes.