Information to Users
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplemental Figure 1. Vimentin
Double mutant specific genes Transcript gene_assignment Gene Symbol RefSeq FDR Fold- FDR Fold- FDR Fold- ID (single vs. Change (double Change (double Change wt) (single vs. wt) (double vs. single) (double vs. wt) vs. wt) vs. single) 10485013 BC085239 // 1110051M20Rik // RIKEN cDNA 1110051M20 gene // 2 E1 // 228356 /// NM 1110051M20Ri BC085239 0.164013 -1.38517 0.0345128 -2.24228 0.154535 -1.61877 k 10358717 NM_197990 // 1700025G04Rik // RIKEN cDNA 1700025G04 gene // 1 G2 // 69399 /// BC 1700025G04Rik NM_197990 0.142593 -1.37878 0.0212926 -3.13385 0.093068 -2.27291 10358713 NM_197990 // 1700025G04Rik // RIKEN cDNA 1700025G04 gene // 1 G2 // 69399 1700025G04Rik NM_197990 0.0655213 -1.71563 0.0222468 -2.32498 0.166843 -1.35517 10481312 NM_027283 // 1700026L06Rik // RIKEN cDNA 1700026L06 gene // 2 A3 // 69987 /// EN 1700026L06Rik NM_027283 0.0503754 -1.46385 0.0140999 -2.19537 0.0825609 -1.49972 10351465 BC150846 // 1700084C01Rik // RIKEN cDNA 1700084C01 gene // 1 H3 // 78465 /// NM_ 1700084C01Rik BC150846 0.107391 -1.5916 0.0385418 -2.05801 0.295457 -1.29305 10569654 AK007416 // 1810010D01Rik // RIKEN cDNA 1810010D01 gene // 7 F5 // 381935 /// XR 1810010D01Rik AK007416 0.145576 1.69432 0.0476957 2.51662 0.288571 1.48533 10508883 NM_001083916 // 1810019J16Rik // RIKEN cDNA 1810019J16 gene // 4 D2.3 // 69073 / 1810019J16Rik NM_001083916 0.0533206 1.57139 0.0145433 2.56417 0.0836674 1.63179 10585282 ENSMUST00000050829 // 2010007H06Rik // RIKEN cDNA 2010007H06 gene // --- // 6984 2010007H06Rik ENSMUST00000050829 0.129914 -1.71998 0.0434862 -2.51672 -

Upregulation of Peroxisome Proliferator-Activated Receptor-Α And
Upregulation of peroxisome proliferator-activated receptor-α and the lipid metabolism pathway promotes carcinogenesis of ampullary cancer Chih-Yang Wang, Ying-Jui Chao, Yi-Ling Chen, Tzu-Wen Wang, Nam Nhut Phan, Hui-Ping Hsu, Yan-Shen Shan, Ming-Derg Lai 1 Supplementary Table 1. Demographics and clinical outcomes of five patients with ampullary cancer Time of Tumor Time to Age Differentia survival/ Sex Staging size Morphology Recurrence recurrence Condition (years) tion expired (cm) (months) (months) T2N0, 51 F 211 Polypoid Unknown No -- Survived 193 stage Ib T2N0, 2.41.5 58 F Mixed Good Yes 14 Expired 17 stage Ib 0.6 T3N0, 4.53.5 68 M Polypoid Good No -- Survived 162 stage IIA 1.2 T3N0, 66 M 110.8 Ulcerative Good Yes 64 Expired 227 stage IIA T3N0, 60 M 21.81 Mixed Moderate Yes 5.6 Expired 16.7 stage IIA 2 Supplementary Table 2. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of an ampullary cancer microarray using the Database for Annotation, Visualization and Integrated Discovery (DAVID). This table contains only pathways with p values that ranged 0.0001~0.05. KEGG Pathway p value Genes Pentose and 1.50E-04 UGT1A6, CRYL1, UGT1A8, AKR1B1, UGT2B11, UGT2A3, glucuronate UGT2B10, UGT2B7, XYLB interconversions Drug metabolism 1.63E-04 CYP3A4, XDH, UGT1A6, CYP3A5, CES2, CYP3A7, UGT1A8, NAT2, UGT2B11, DPYD, UGT2A3, UGT2B10, UGT2B7 Maturity-onset 2.43E-04 HNF1A, HNF4A, SLC2A2, PKLR, NEUROD1, HNF4G, diabetes of the PDX1, NR5A2, NKX2-2 young Starch and sucrose 6.03E-04 GBA3, UGT1A6, G6PC, UGT1A8, ENPP3, MGAM, SI, metabolism -

Identification of a Novel Nucleobase-Ascorbate Transporter
bioRxiv preprint doi: https://doi.org/10.1101/287870; this version posted December 7, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. 1 Identification of a novel Nucleobase-Ascorbate 2 Transporter family member in fish and amphibians 3 Diogo Oliveira*,1, André M. Machado*,1, Tiago Cardoso1, Mónica Lopes-Marques1, L. Filipe 4 C. Castro1,2● and Raquel Ruivo1● 5 1CIIMAR – Interdisciplinary Centre of Marine and Environmental Research, U. Porto – University 6 of Porto, Porto, Portugal 7 2Department of Biology, Faculty of Sciences, U. Porto - University of Porto, Portugal 8 *Equal contribution 9 ●Corresponding authors at: CIIMAR, Terminal de Cruzeiros do Porto de Leixões, Av. General 10 Norton de Matos s/n, 4450-208 Matosinhos, Portugal. Tel.: +351 223 401 831 11 12 Running title: Novel uric acid transporter in fish and amphibians 13 14 15 bioRxiv preprint doi: https://doi.org/10.1101/287870; this version posted December 7, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. 16 Abstract:Nucleobase-Ascorbate Transporter (NAT) family includes ascorbic acid, nucleobases 17 and uric acid transporters: with a broad evolutionary distribution. In vertebrates, four members 18 have been previously recognized, the ascorbate transporters Slc23a1 and Slc3a2, the nucleobase 19 transporter Slc23a4 and an orphan transporter Slc23a3. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Mouse Slc22a2 Knockout Project (CRISPR/Cas9)
https://www.alphaknockout.com Mouse Slc22a2 Knockout Project (CRISPR/Cas9) Objective: To create a Slc22a2 knockout Mouse model (C57BL/6N) by CRISPR/Cas-mediated genome engineering. Strategy summary: The Slc22a2 gene (NCBI Reference Sequence: NM_013667 ; Ensembl: ENSMUSG00000040966 ) is located on Mouse chromosome 17. 11 exons are identified, with the ATG start codon in exon 1 and the TAA stop codon in exon 11 (Transcript: ENSMUST00000046959). Exon 2 will be selected as target site. Cas9 and gRNA will be co-injected into fertilized eggs for KO Mouse production. The pups will be genotyped by PCR followed by sequencing analysis. Note: Mice homozygous for a knockout allele are viable and fertile and display no obvious phenotypic abnormalities. No significant defects in the renal secretion of a model organic cation are observed. Exon 2 starts from about 25.02% of the coding region. Exon 2 covers 6.27% of the coding region. The size of effective KO region: ~104 bp. The KO region does not have any other known gene. Page 1 of 8 https://www.alphaknockout.com Overview of the Targeting Strategy Wildtype allele 5' gRNA region gRNA region 3' 1 2 11 Legends Exon of mouse Slc22a2 Knockout region Page 2 of 8 https://www.alphaknockout.com Overview of the Dot Plot (up) Window size: 15 bp Forward Reverse Complement Sequence 12 Note: The 2000 bp section upstream of Exon 2 is aligned with itself to determine if there are tandem repeats. Tandem repeats are found in the dot plot matrix. The gRNA site is selected outside of these tandem repeats. -
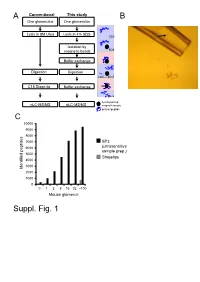
Suppl. Fig. 1 A
A Conventional This study B One glomerulus One glomerulus Lysis in 8M Urea Lysis in 4% SDS SDS Isolation by magnetic beads SDS Buffer exchange Digestion Digestion Try LysC pH>7 C18 Stage tip Buffer exchange pH<4 functionalized nLC-MS/MS nLC-MS/MS magnetic beads protein/peptide C 10000 9000 8000 7000 SP3 6000 (ultrasensitive sample prep.) 5000 Stagetips 4000 3000 Identified peptides 2000 1000 0 0 1 2 8 16 32 ~100 Mouse glomeruli Suppl. Fig. 1 S1. Sample preparation methods. A. Scheme of conventional (C18 Stage Tip) and ultrasensitive proteomic sample preparation methods. B. Manual microdissection and isolation of single glomeruli. The arrow indicates a single isolated glomerulus subjected to proteome analysis. The tip capacity is 10µl. C. Stagetips, a conventional proteomic sample preparation method, is compared with ultrasensitive (SP3) proteome analysis. The indicated amounts of mouse glomeruli were prepared by the respective sample preparation protocols, and peptide numbers after nLC-MS/MS are plotted (all FDR<0.01). A 100 1 Tubule B 80 60 40 Intensity CCD 20 40 0 100 20 S1 TAL 80 0 Tubule proximal tubule Loading... 60 0 Intensity 40 S2 20 -20 control Component 2 (22.5%) S3 0 0 10 20 30 40 50 60 -40 -20 0 20 40 Time (min) Component 2 (47.1%) C D 6000 Mouse proximal tubule 5000 8 4000 3000 7 (iBAQ) Loading... 10 2000 log 6 Number of peptides 1000 0 5 Control mouse mouse mouse human proximal mTAL CCD proximal 0 1500 tubule tubule Rank protein (S1) (S1) R=0.7 E F 8 ACTB Human proximal tubule 8 7 SLC9A3R1 ATP1A1 SLC3A2 SLC25A5 ATP1B1 7 SLC5A2 SLC25A3 CUBN CLTC SLC27A2 SLC5A12 LRP2 6 SLC25A13 SLC25A10 SLC5A1 SLC22A6 6 SLC25A4 Loading.. -

The Concise Guide to Pharmacology 2019/20
Edinburgh Research Explorer THE CONCISE GUIDE TO PHARMACOLOGY 2019/20 Citation for published version: Cgtp Collaborators 2019, 'THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Transporters', British Journal of Pharmacology, vol. 176 Suppl 1, pp. S397-S493. https://doi.org/10.1111/bph.14753 Digital Object Identifier (DOI): 10.1111/bph.14753 Link: Link to publication record in Edinburgh Research Explorer Document Version: Publisher's PDF, also known as Version of record Published In: British Journal of Pharmacology General rights Copyright for the publications made accessible via the Edinburgh Research Explorer is retained by the author(s) and / or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Take down policy The University of Edinburgh has made every reasonable effort to ensure that Edinburgh Research Explorer content complies with UK legislation. If you believe that the public display of this file breaches copyright please contact [email protected] providing details, and we will remove access to the work immediately and investigate your claim. Download date: 28. Sep. 2021 S.P.H. Alexander et al. The Concise Guide to PHARMACOLOGY 2019/20: Transporters. British Journal of Pharmacology (2019) 176, S397–S493 THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Transporters Stephen PH Alexander1 , Eamonn Kelly2, Alistair Mathie3 ,JohnAPeters4 , Emma L Veale3 , Jane F Armstrong5 , Elena Faccenda5 ,SimonDHarding5 ,AdamJPawson5 , Joanna L -
And Mir183 in Mir183/96 Dko Mutant Mice (Top) And
Supplementary Information Appendix Figure S1. Expression of Mir96 , Mir182 and Mir183 in Mir183/96 dko mutant mice (top) and Mir182 ko mutant mice (bottom), relative to Mir99a , which is expressed in cochlear sensory epithelium. Homozygote (red; right bars) and heterozygote (blue; middle bars) expression levels have been normalised to expression in the wildtype (green; left bars). Mir183/96 dko : wildtype n=7, heterozygote n=5, homozygote n=6. Mir182 ko : wildtype n=4, heterozygote n=4, homozygote n=4. Error bars are standard deviation (* = P < 0.05, ** = P < 0.01). All p-values were calculated using the Wilcoxon rank sum test. For Mir183/96 dko heterozygotes, Mir96 p=0.002525; Mir182 p=0.6389; Mir183 p=0.002525. For Mir183/96 dko homozygotes, Mir96 p=0.002067; Mir182 p=0.1014; Mir183 p=0.002067. For Mir182 ko heterozygotes, Mir96 p=0.05714; Mir182 p=0.3429; Mir183 p=0.3429. For Mir182 ko homozygotes, Mir96 p=1; Mir182 p=0.02652; Mir183 p=0.05714. 67 68 Appendix Figure S2. Individual ABR thresholds of wildtype, heterozygous and homozygous Mir183/96 dko mice at all ages tested. Number of mice of each genotype tested at each age is shown on the threshold plot. 69 70 Appendix Figure S3. Individual ABR thresholds of wildtype, heterozygous and homozygous Mir182 ko mice at all ages tested. Number of mice of each genotype tested at each age is shown on the threshold plot. 71 Appendix Figure S4. Mean ABR waveforms at 12kHz, shown at 20dB (top) and 50dB (bottom) above threshold (sensation level, SL) ± standard deviation, at four weeks old. -

Towards the Elucidation of Orphan Lysosomal Transporters Quentin Verdon
Towards the Elucidation of Orphan Lysosomal Transporters Quentin Verdon To cite this version: Quentin Verdon. Towards the Elucidation of Orphan Lysosomal Transporters. Cancer. Université Paris Saclay (COmUE), 2016. English. NNT : 2016SACLS144. tel-01827233 HAL Id: tel-01827233 https://tel.archives-ouvertes.fr/tel-01827233 Submitted on 2 Jul 2018 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. NNT : 2016SACLS144 THESE DE DOCTORAT DE L’UNIVERSITE PARIS-SACLAY PREPAREE A L’UNIVERSITE PARIS-SUD ECOLE DOCTORALE N°568 BIOSIGNE | Signalisations et réseaux intégratifs en biologie Spécialité de doctorat : aspects moléculaires et cellulaires de la biologie Par Mr Quentin Verdon Towards the elucidation of orphan lysosomal transporters: several shots on target and one goal Thèse présentée et soutenue à Paris le 29/06/2016 » : Composition du Jury : Mr Le Maire Marc Professeur, Université Paris-Sud Président Mr Birman Serge Directeur de recherche, CNRS Rapporteur Mr Murray James Assistant professor, Trinity college Dublin Rapporteur Mr Goud Bruno Directeur de recherche, CNRS Examinateur Mr Gasnier Bruno Directeur de recherche, CNRS Directeur de thèse Mme Sagné Corinne Chargée de recherche, INSERM Co-directeur de thèse Table of contents Remerciements (acknowledgements) 6 Abbreviations 7 Abstracts 10 Introduction 12 1 Physiology of lysosomes 12 1.1 Discovery and generalities 12 1.2 Degradative function 13 1.3. -

Toxicological Profile for Nitrate and Nitrite
NITRATE AND NITRITE 29 3. HEALTH EFFECTS 3.1 INTRODUCTION The primary purpose of this chapter is to provide public health officials, physicians, toxicologists, and other interested individuals and groups with an overall perspective on the toxicology of nitrate and nitrite. It contains descriptions and evaluations of toxicological studies and epidemiological investigations and provides conclusions, where possible, on the relevance of toxicity and toxicokinetic data to public health. A glossary and list of acronyms, abbreviations, and symbols can be found at the end of this profile. 3.2 DISCUSSION OF HEALTH EFFECTS BY ROUTE OF EXPOSURE To help public health professionals and others address the needs of persons living or working near hazardous waste sites, the information in this section is organized first by route of exposure (inhalation, oral, and dermal) and then by health effect (e.g., death, systemic, immunological, neurological, reproductive, developmental, and carcinogenic effects). These data are discussed in terms of three exposure periods: acute (14 days or less), intermediate (15–364 days), and chronic (365 days or more). Levels of significant exposure for each route and duration are presented in tables and illustrated in figures. The points in the figures showing no-observed-adverse-effect levels (NOAELs) or lowest- observed-adverse-effect levels (LOAELs) reflect the actual doses (levels of exposure) used in the studies. LOAELs have been classified into "less serious" or "serious" effects. "Serious" effects are those that evoke failure in a biological system and can lead to morbidity or mortality (e.g., acute respiratory distress or death). "Less serious" effects are those that are not expected to cause significant dysfunction or death, or those whose significance to the organism is not entirely clear. -

Taqman® Openarray® Pharmacogenomics (Pgx) Panel
PRODUCT OVERVIEW TaqMan® OpenArray® Pharmacogenomics (PGx) Panel TaqMan® OpenArray® Pharmacogenomics (PGx) Panel Genotyping analysis of drug metabolism enzymes and associated transport proteins The TaqMan® OpenArray® Trusted TaqMan® performance samples, studies, and labs. Assays Pharmacogenomics (PGx) Panel is Each TaqMan® DME Genotyping were selected for optimal relevance a powerful tool to help researchers Assay contains two allele-specific to current pharmacogenomics study human genetic variation probes and a primer pair to detect studies and organized for a simplified in relation to drug action and its the specific SNP target. Both the workflow. potential application to medical probes and primers uniquely align ® ® treatment. within the genome, enabling the The TaqMan OpenArray PGx TaqMan® genotyping technology to Panel provides valuable data for ® ® The TaqMan OpenArray PGx provide superior specificity. It is this the study of drug interactions in Panel was developed for quick and specificity that allows these assays several research areas. Targeted easy screening of known high- to detect targets residing in highly genes relate to areas of study such value target genes associated with homologous gene families that may as cardiovascular (CYP2D6, CYP2C19, drug metabolism enzymes and include pseudogenes. TaqMan® Drug NAT1, NAT2), analgesics (CYP2C9, associated transport proteins. Metabolism Enzyme Genotyping CYP2D6), rheumatology (CYP2C9, The panel consists of 158 drug Assays were developed using a high TPMT), neurology (CYP2C19, CYP2D6), metabolism enzyme (DME) assays level of bioinformatics and wet-lab and musculoskeletal (CYP2C19). A derived from the PharmaADME stringency. All assays have passed total of 29 genes are covered across Core Marker Set (Table 1). The performance tests involving 180 158 unique assays. -

Role of Membrane Transportors in Cisplatin Induced Nephrotoxicity
Hematology & Medical Oncology Review Article ISSN: 2398-8495 Role of membrane transportors in cisplatin induced nephrotoxicity Sakshi Rajput and Gaaminepreet Singh* Department of pharmacology, ISF College of Pharmacy, Moga, Punjab, India Abstract Transporters are important mediators of specific cellular uptake and thus, not only for effects, but also for side effects, metabolism, and excretion of many drugs such as cisplatin. Cisplatin is a potent cytostatic drug, whose use is limited by its severe acute and chronic nephro-, oto-, and peripheral neurotoxicity. For this reason, other platinum derivatives, such as carboplatin and oxaliplatin, with less toxicity but still with antitumoral action have been developed. Several transporters, which are expressed on the cell membranes, have been associated with cisplatin transport across the plasma membrane and across the cell: the copper transporter 1 (Ctr1), the copper transporter 2 (Ctr2), the P-type copper-transporting ATPases ATP7A and ATP7B, the organic cation transporter 2 (OCT2), and the multidrug extrusion transporter 1 (MATE1). Some of these transporters are also able to accept other platinum derivatives as substrate. Since membrane transporters display a specific tissue distribution, they can be important molecules that mediate the entry of platinum derivatives in target and also non-target cells possibly mediating specific effects and side effects of the chemotherapeutic drug. this paper summarizes the literature on toxicities of cisplatin compared to that of carboplatin and oxaliplatin and the interaction of these platinum derivatives with membrane transporters. Abbreviation: Ctr1: Copper Transporter 1; Ctr2: Copper Trans- antitumor drugs in the world [6]. porter 2; OCT2: Organic Cation Transporter 2; MATE1: Multidrug and Cisplatin was the first platinum-based drug that revolutionized the Extrusion Transporter 1; FDA: Food and Drug Administration; DNA: treatment of neoplastic diseases.